 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 53(4); 2019 > Article
-
Original Article
Progressive Familial Intrahepatic Cholestasis in Korea: A Clinicopathological Study of Five Patients -
Hyo Jeong Kang1,*
 , Soon Auck Hong2,*, Seak Hee Oh3, Kyung Mo Kim3, Han-Wook Yoo3, Gu-Hwan Kim4, Eunsil Yu,1,5
, Soon Auck Hong2,*, Seak Hee Oh3, Kyung Mo Kim3, Han-Wook Yoo3, Gu-Hwan Kim4, Eunsil Yu,1,5 -
Journal of Pathology and Translational Medicine 2019;53(4):253-260.
DOI: https://doi.org/10.4132/jptm.2019.05.03
Published online: May 16, 2019
11Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
22Department of Pathology, Soonchunhyang University Cheonan Hospital, Cheonan, Korea
3Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
4Medical Genetics Center, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
5Asan Liver Center, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- Corresponding Author Eunsil Yu, MD, PhD Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea Tel: +82-2-3010-4552 Fax: +82-2-472-7898 E-mail: esyu@amc.seoul.kr
- *Hyo Jeong Kang and Soon Auck Hong contributed equally to this work.
© 2019 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background
- Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of autosomal recessive liver diseases that present as neonatal cholestasis. Little is known of this disease in Korea.
-
Methods
- The records of five patients histologically diagnosed with PFIC, one with PFIC1 and four with PFIC2, by liver biopsy or transplant were reviewed, and ATP8B1 and ABCB11 mutation status was analyzed by direct DNA sequencing. Clinicopathological characteristics were correlated with genetic mutations.
-
Results
- The first symptom in all patients was jaundice. Histologically, lobular cholestasis with bile plugs was the main finding in all patients, whereas diffuse or periportal cholestasis was identified only in patients with PFIC2. Giant cells and ballooning of hepatocytes were observed in three and three patients with PFIC2, respectively, but not in the patient with PFIC1. Immunostaining showed total loss of bile salt export pump in two patients with PFIC2 and focal loss in two. Lobular and portal based fibrosis were more advanced in PFIC2 than in PFIC1. ATP8B1 and ABCB11 mutations were identified in one PFIC1 and two PFIC2 patients, respectively. One PFIC1 and three PFIC2 patients underwent liver transplantation (LT). At age 7 months, one PFIC2 patient was diagnosed with concurrent hepatocellular carcinoma and infantile hemangioma in an explanted liver. The patient with PFIC1 developed steatohepatitis after LT. One patient showed recurrence of PFIC2 after 10 years and underwent LT.
-
Conclusions
- PFIC is not rare in patients with neonatal cholestasis of unknown origin. Proper clinicopathologic correlation and genetic testing can enable early detection and management.
- Ethical approval
- This study was approved by the Institutional Review Board of Asan Medical Center, which waived the requirement for informed consent (IRB No. 2017-0703).
- Patients and samples
- From 2008 to 2014, five patients were diagnosed with PFIC at Asan Medical Center, Seoul, Republic of Korea. Diagnosis was based on clinical findings, histological and ultrastructural features of liver biopsy or liver transplantation (LT) samples, and mutation profiles. Clinical information, including age at onset, sex, chief complaints, and laboratory data, were retrieved from patients’ electronic medical records. Informed consent for liver biopsy and DNA analysis in all patients was obtained from the patients’ parents. No patients were lost to follow-up and the median follow-up time was 53 months (range, 36 to 102 months).
- Histopathological analysis
- Fifteen specimens from four liver biopsies, eight posttransplant liver biopies, and three explanted livers were histopathologically analyzed. All specimens were fixed in 10% neutral formalin and processed for hematoxylin-eosin and Masson trichrome staining to analyze histology and the stage of fibrosis. Specimens for electron microscropy were fixed in 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) for 48 hours, and postfixed in 1% osmium tetroxide. Ultrathin sections were cut, stained with uranyl acetate, and examined using a transmission electron microscope.
- Samples were immunohistochemically stained with antibodies to cytokeratin 19 (1:200, DAKO, Glostrup, Denmark) and BSEP (1:1,000, Sigma Aldrich, St. Louis, MO, USA) to assess bile duct damage and BSEP protein expression, respectively, using a Benchmark XT auto immunostainer (Ventana Medical System, Tucson, AZ, USA) with OptiView DAB IHC Detection kit (Ventana Medical System) according to the manufacturer’s instructions. Histopathological features were carefully reviewed by three pathologists (E.Y., S.A.H., and H.J.K.) specializing in liver diseases, and the absence or presence of ballooning degeneration, giant cell transformation, and ductular reaction, as well as the extent of bile plugs, was determined. Lobular and portal based fibrosis were also analyzed, according to the Korean Society of Pathologists Scoring System [8,9]. Lobular fibrosis was graded as negative (no perivenular fibrosis), mild (perivenular fibrosis), moderate (perivenular and sinusoidal fibrosis), or severe (diffuse perivenular and sinusoidal fibrosis). Portal fibrosis was graded as no portal fibrosis, periportal fibrosis, septal fibrosis and cirrhosis.
- Genetic test
- Genomic DNA was isolated from peripheral blood leukocytes using Gentra Puregene blood kits (Qiagen, Hilden, Germany). Twenty-seven exons and exon-intron boundaries of ATP8B1 and 28 exons and exon-intron boundaries of ABCB11 were amplified by polymerase chain reaction (PCR) using GoTaq Polymerase (Promega, Madison, WI, USA), and the products were directly sequenced using BigDye Terminator kits v.3.1 (Applied Biosystems/Life Technologies, Waltham, MA, USA). The PCR products were electrophoresed using an ABI3130x1 Genetic Analyzer (Applied Biosystems/Life Technologies), and DNA sequences were compared with GenBank (http://www.ncbi.nlm.nih.gov) reference DNA sequences: NT_0025028.14 and NT_005403.17 for genomic sequences of ATP8B1 and ABCB11, respectively; and NM_005603.4 and NM_003742.2 for ATP8B1 and ABCB11 mRNAs, respectively. In silico prediction of the effects of missense variants in ATP8B1 and ABCB11 were performed using Poly-Phen-2 (http://genetics.bwh.harvard.edu/pph2) and Sorting Intolerance from Tolerant (SIFT; http://sift.jcvi.org).
MATERIALS AND METHODS
- Clinical and laboratory findings
- The clinical characteristics of the five patients with PFIC are listed in Table 1. One was diagnosed with PFIC1 and four with PFIC2. Four were female and one was male. Age at PFIC onset was birth in one patient with PFIC1 and three with PFIC2, and 45 days in a fourth patient with PFIC2. Four patients were Korean and one was Arabian. The Arabian girl had been previously diagnosed with PFIC2 and underwent LT at another hospital. Jaundice was common to all five patients (100%). Only one PFIC2 patient had pruritus in addition to cholestasis. The median total bilirubin and direct bilirubin concentrations at the time of diagnosis were 19.6 mg/dL (range, 10.0 to 24.4 mg/dL; normal range, 0.2 to 1.2 mg/dL) and 15.0 mg/dL (range, 5.6 to 21.0; normal range, < 0.5 mg/dL), respectively. All patients had low or normal GGT (median, 30.0 mg/dL; range, 15.0 to 57.0 mg/dL; normal range, 8 to 61 mg/dL). Four patients, one with PFIC1 and three with PFIC2, underwent LT, followed by allograft liver biopsies performed two or three times.
- Histopathological findings
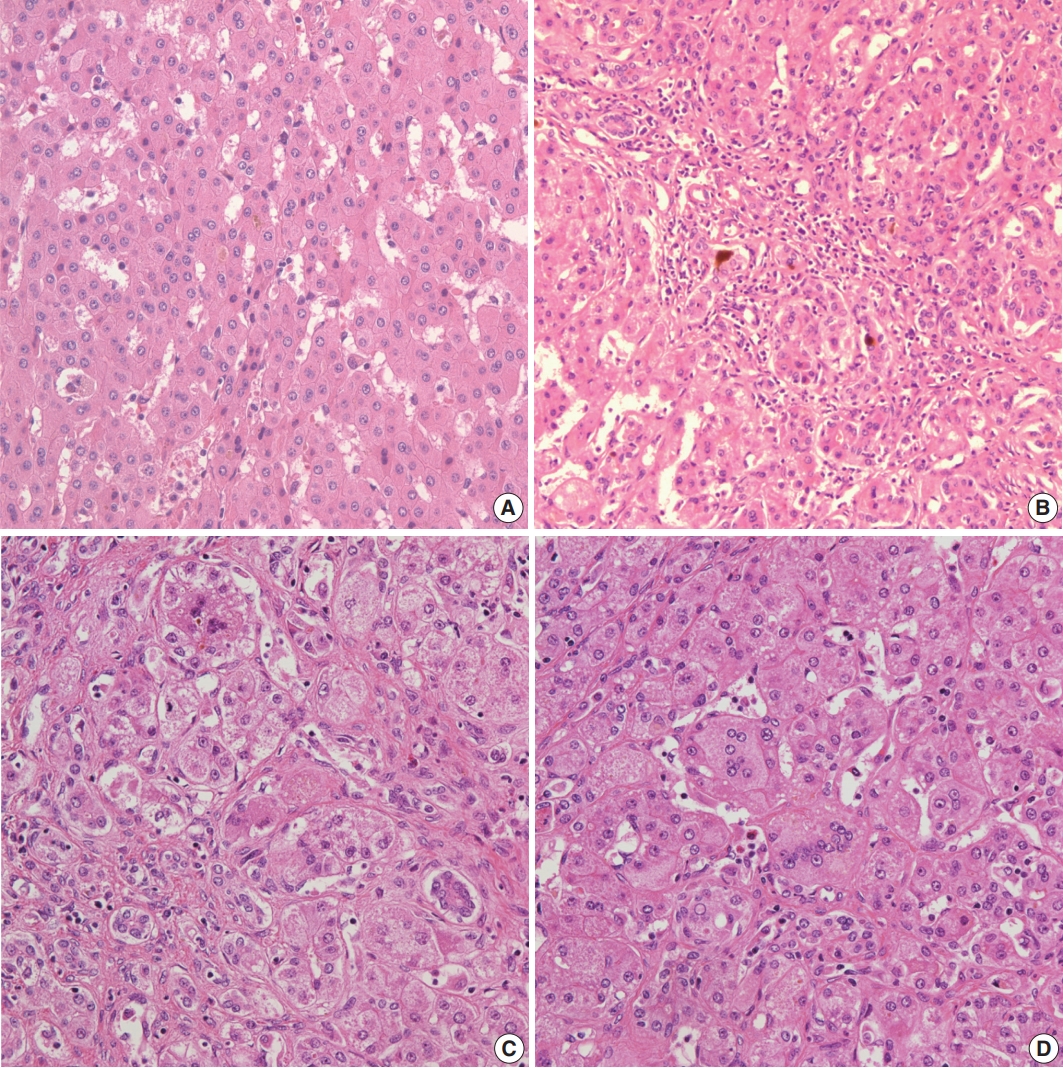
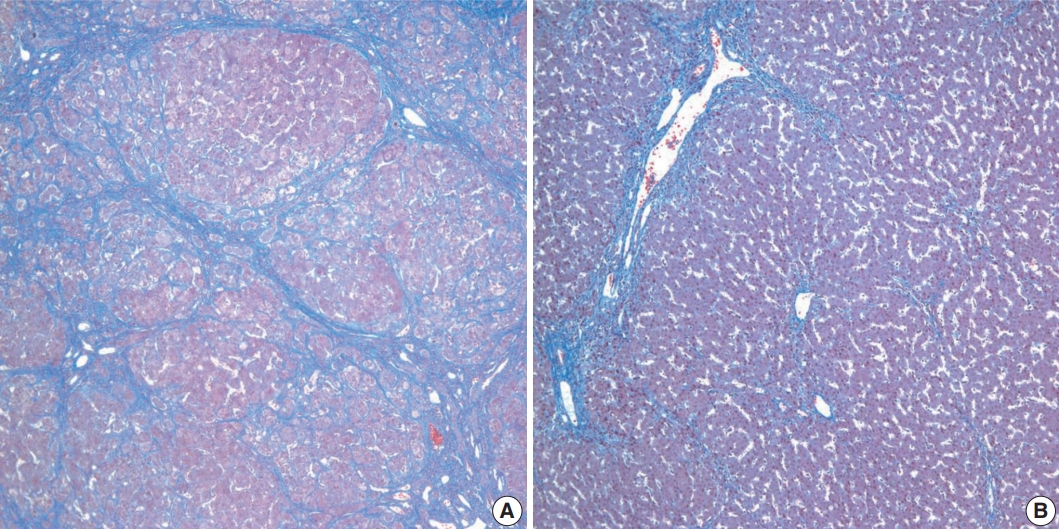
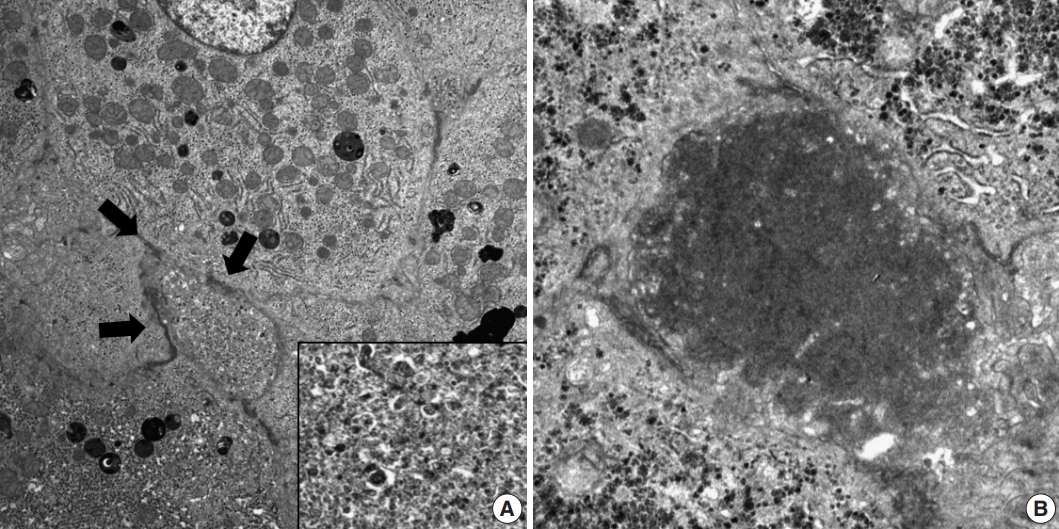
- Histopathological features of the five patients with PFIC are shown in Table 2. All had intrahepatic cholestasis, but the patterns of cholestasis were different in patients with PFIC1 and PFIC2. The main finding in the patient with PFIC1 was bland lobular cholestasis with bile plugs (Fig. 1A), whereas the main findings in the patients with PFIC2 were canalicular, periportal, and/or cholangiolar cholestasis (Fig. 1B). Ballooning degeneration of hepatocytes (Fig. 1C) and giant cell transformation (Fig. 1D) were observed in three and three patients with PFIC2, respectively. Small cell changes in hepatocytes were observed only in the PFIC1 patient (Fig. 1A). Lobular activity ranged from none to severe, whereas portal activity was mild in the four patients with PFIC2 (75%). Ductular reactions were observed in the one patient with PFIC1 and in four with PFIC2. The stage of fibrosis varied in patients with PFIC2, with one having cirrhosis, two having periportal fibrosis, and one having no portal fibrosis. The PFIC1 patient had periportal fibrosis. Severe lobular fibrosis was observed in three PFIC2 patients (Fig. 2A), and mild fibrosis in one PFIC1 and one PFIC2 patient (Fig. 2B). Electron microscopy showed coarse and granular bile, called Byler’s bile, in the one PFIC1 patient (Fig. 3A), and amorphous bile in four patients with PFIC2 (Fig. 3B).
- BSEP immunostaining
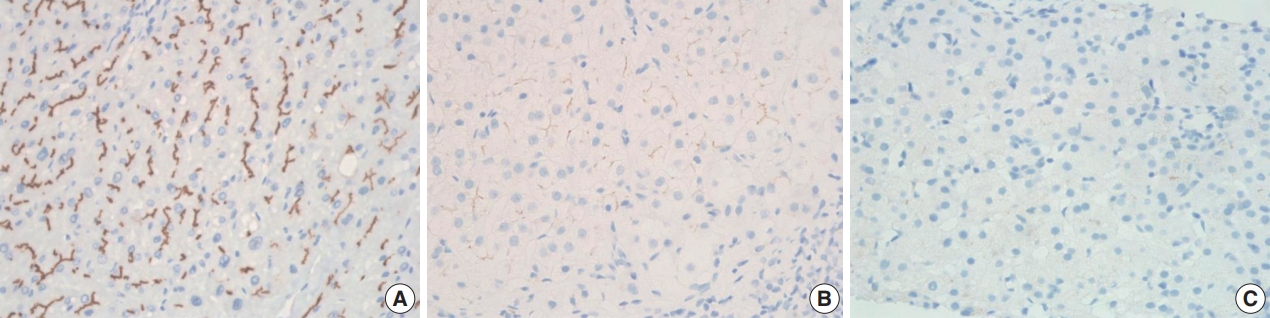
- BSEP immunohistochemical staining in patient with one PFIC1 showed normal canalicular expression (Fig. 4A). PFIC2 showed focal loss of BSEP expression in two patients (Fig. 4B) and total loss of BSEP expression in two patients (Fig. 4C), with the latter having an ABCB11 gene mutation.
- Genetic mutation analysis
- Genetic testing (Table 3) showed a nonsense mutation, c.1753G > T (p.E585*), in ATP8B1 in the one patient with PFIC1. Genetic data were available from all PFIC2 patients. Two patients had mutations in ABCB11, with one having a missense mutation, c.1907A > G (p.E636G), and the other having a nonsense mutation, c.1416T > A (p.Y472*). No mutations on ABCB11 gene were identified in two cases of PFIC2. In addition, the PFIC1 patient had a missense mutation, c.2246T > C (p.L749P) (c.2246T > C) in ATP8B1, and one PFIC2 patient had a missense mutation, c.2594C > T (p.A865V) (c.2594C > T) in ABCB11, with Poly-Phen-2 (http://genetics.bwh.harvard.edu/pph2) and SIFT (http://sift.jcvi.org) predicting that these missense mutations would lead to functional derangements of their respective proteins. These novel mutations could not be validated to exclude polymorphisms.
- Clinicopathological course
- An abdominal computed tomography scan in one PFIC2 patient before LT showed multiple enhancing nodules and masses in both lobes of the liver. These were histologically found to be hepatocellular carcinomas and multiple infantile hemangiomas. Recurrence following LT was identified in the one Arabian PFIC2 patient. This patient experienced malnutrition and intractable jaundice 12 years after LT. Laboratory tests showed that, despite markedly elevated asparate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, and direct bilirubin concentrations, her GGT stayed in the normal range. Liver biopsy showed canalicular and periportal bile plugs, as well as diffuse giant cell and ballooning changes in hepatic lobules. Masson trichrome staining showed periportal fibrosis, and BSEP immunostaining showed complete loss of BSEP protein from the canalicular membrane.
- The one patient diagnosed with PFIC1 did not show the expected recovery of AST and ALT concentrations after LT. A posttransplant liver biopsy showed severe fatty changes, ballooning degeneration, and mild lobular activity accompanied by sinusoidal and perivenular fibrosis. The final diagnosis was steatohepatitis, a frequent complication of LT in PFIC1 patients. One PFIC2 patient each developed acute cellular rejection and ischemic liver damage. All patients were alive at the last follow-up.
RESULTS
- Although PFIC is a relatively well-documented disease worldwide, there have been only two case reports on PFIC in Korean patients. Identification of a mutation in an associated gene is not mandatory for diagnosis of PFIC, but is an important diagnostic tool [1]. However, clinical segmentation remains useful because, although PFIC has been reclassified according to molecular criteria, genetic testing is not always readily available. To our knowledge, this is the first study describing overall features of patients in Korea with PFIC.
- PFIC is a heterogeneous group of liver diseases, but laboratory findings are similar in PFIC1 and PFIC2 patients, such as low GGT despite direct hyperbilirubinemia. These results suggest that hyperbilirubinemia in PFIC is associated with bile excretion by hepatocytes. By contrast, common forms of neonatal cholestasis are characterized by increased GGT, usually from damage to the bile ducts caused by the detergent effect of bile salts [10,11]. Although PFIC is unfamiliar to general pediatricians and pathologists, it is not a rare disease, accounting for 10%–15% of patients with neonatal cholestasis of unknown etiology. Knowledge of laboratory findings characteristic of PFIC narrows the differential diagnoses, enabling early diagnosis of PFIC in patients with intrahepatic cholestasis.
- Some histopathologic features differ in PFIC1 and PFIC2 [12]. For example, PFIC1 is characterized by a bland lobular cholestasis with hepatocellular and canalicular patterns, as well as small cell changes in hepatocytes in early life biopsies [13]. Because our PFIC1 patient showing small cell change was 2 years old at the time of LT, the presence of small cell change is not limited to infancy but appears over time. Giant cell transformation of hepatocytes and ballooning change with cholestasis were major histologic features in PFIC2 patients, although the amount and distribution of giant cells and ballooning changes vary [14].
- In contrast to PFIC2, abnormalities of the interlobular bile ducts are not observed in PFIC1 [2]. However, ductular reactions may appear in the latter, indicating disease progression [12]. The ductular reaction in our one PFIC1 patient may reflect her relatively older age.
- Liver fibrosis is an early event of PFIC, with the grade and pattern of fibrosis influenced by patient age and type of PFIC. Lobular fibrosis may develop in PFIC1 and PFIC2, whereas portal fibrosis is a predominant feature of PFIC2 [15]. Portal fibrosis may be more associated with ductular reaction and portal pathology of PFIC2.
- Electron microscopy has shown that bile properties differ in PFIC1 and PFIC2. Coarse granular bile, called Byler’s bile, is a distinct characteristic of PFIC1 and was used to diagnose our patient [16]. By contrast, PFIC2 is characterized by nonspecific fine bile located within dilated canaliculi, similar to findings in patients with intrahepatic cholestasis of various etiologies. Electron microscopic examination should therefore be included in the differential diagnosis of pediatric patients with cholestatic liver diseases.
- About 80 and 100 genetic mutations have been identified in PFIC1 and PFIC2 patients, respectively [17,18]. Despite PFICs being autosomal recessive hereditary diseases, compound heterozygous or homozygous mutations may be associated with structural and functional defects. More severe forms of PFIC are likely associated with homozygous frame shift and nonsense mutations, as well as large genomic deletion. By contrast, a milder form of PFIC, benign recurrent intrahepatic cholestasis, is more likely associated with heterozygous missense mutations [19].
- Most point mutations in PFIC1 and PFIC2 are missense, nonsense, and splicing mutations. Structurally abnormal proteins in PFIC1 are due to frame shift (26%), splice site (18%), and nonsense (13%) mutations, as well as large genomic deletion (3%). Functionally deficient proteins with normal structure in PFIC1 are caused by missense mutations (38%) and small inframe deletions (3%) [19]. The PFIC1 patient in the present study was found to have two heterozygous mutations; one nonsense, c.1753G > T (p.E585*), and one missense, c.2246T > C (p.L749P), mutation. Heterozygous mutations have been associated with good prognosis and low penetrance in PFIC [4,13]. However, our PFIC1 patient required an LT at age 26 months, suggesting that heterozygous mutations could not guarantee a benign clinical course in PFIC1 patients.
- We failed to detect ABCB11 mutations in two PFIC2 patients in the present study. Fewer than 10% of PFIC patients have no or monoallelic mutations. Mutations in these patients may be present in regulatory domains, untranslated regions, and introns, which cannot be tested by present methods [20]. Despite the absence of mutations or the presence of a single heterozygous ABCB11 mutation, the diagnostic sensitivity of clinical and pathologic findings with negative BSEP immunostaining is approximately 90% [18]. Therefore, the absence of ABCB11 mutations cannot exclude PFIC2.
- Ursodeoxycholic acid (UDCA) is considered an initial treatment for patients with PFICs [21]. UDCA can resolve symptoms in PFIC1 patients, but PFIC2 patients generally respond poorly, suggesting that UDCA has uncertain effects on the progression of liver disease [22]. In early stages of PFIC, partial biliary diversion that causes an unloading of bile acid may delay LT [23]. LT is the most efficient and last therapeutic option in patients presenting with liver failure [24,25]. The selection criteria for LT candidates do not differ from those in patients with other liver diseases. Major indications for LT include end stage liver disease, concurrent hepatocellular carcinoma, and intractable pruritus with no response to biliary diversion. Although LT results in remission in 75%–100% of patients, regardless of PFIC type, specific complications and relapse of disease should be carefully considered prior to LT [24-26]. Patients with PFIC1 may experience exacerbation of extrahepatic manifestations, including chronic diarrhea and liver steatosis [27,28]. For example, our one PFIC1 patient experienced rapid development of steatohepatitis not observed in the explant liver.
- In summary, PFIC is not infrequently encountered in practice. Early detection and management should be based on proper clinicopathologic correlation and mutation analysis. Because it is more cost-effective, BSEP immunostaining should be performed prior to genetic testing to determine the type of PFIC. Mutation analysis of the ABCB11 gene should be performed in patients with portal tract abnormalities and reduction or loss of BSEP expression on immunostaining. In addition, electron microscopy should always be considered for the diagnosis and typing of PFIC.
DISCUSSION
Author Contributions
Conceptualization: EY, SAH, HJK.
Data curation: SAH, SHO, KMK, HWY.
Methodology: SHO, KMK, HWY.
Supervision: EY.
Visualization: HJK, SAH, GHK.
Writing—original draft: HJK, SAH.
Writing—review & editing: EY
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
| Patient No. | PFIC type | Sex/Age at onset | Initial symptom | AST (IU/L) | ALT (IU/L) | GGT (mg/dL) | ALP (IU/L) | TB (mg/dL) | DB (mg/dL) | Concomitant tumor | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | F/Birth | Jaundice | 282 | 152 | 32 | 356 | 24.4 | 21.0 | HCC, infantile hemangioma | LT |
| 2 | 2 | M/45 days | Jaundice | 72 | 65 | 15 | 594 | 10 | 5.6 | Absent | UDCA |
| 3 | 2 | F/Birth | Jaundice | 43 | 20 | 57 | 576 | 10.6 | 7.6 | Absent | LT |
| 4a | 2 | F/Birth | Jaundice | 468 | 553 | 30 | 402 | 20.0 | 15.0 | Absent | LT |
| 5 | 1 | F/Birth | Jaundice | 69 | 42 | 16 | 824 | 19.6 | 19.0 | Absent | LT |
PFIC, progressive familiar intrahepatic cholestasis; AST, asparate aminotransferase; ALT, alanine aminotransferase; GGT, gamma-glutamyl transpeptidase; ALP, alkaline phosphatase; TB, total bilirubin; DB, direct bilirubin; F, female; HCC, hepatocellular carcinoma; LT, liver transplantation; M, male; UDCA, urodeoxycholic acid.
aArabian.
| Pathologic parameter | Case 1 (PFIC2) | Case 2 (PFIC2) | Case 3 (PFIC2) | Case 4a (PFIC2) | Case 5 (PFIC1) |
|---|---|---|---|---|---|
| Specimen type | Biopsy (× 1) | Biopsy (×1) | Biopsy (× 1) | Posttansplant Bx (×3) | Biopsy (× 1) |
| Explanted liver (× 1) | Explanted liver (× 1) | Explanted liver (× 1) | |||
| Posttansplant Bx (× 1) | Posttransplant Bx (4) | ||||
| Bile plugs | Canalicular/Periportal/Cholangiolar | Canalicular | Canalicular/Periportal | Canalicular/Periportal | Canalicular |
| Lobular activity | Absent | Absent | Severe | Mild | Absent |
| Giant cell change | Present | Absent | Present | Present | Absent |
| Ballooning change | Present | Absent | Present | Present | Absent |
| Small cell change | Absent | Absent | Absent | Absent | Present |
| Portal activity | Mild | Absent | Mild | Mild | Mild |
| Ductular reaction | Present | Present | Present | Present | Present |
| Lobular-based fibrosis | Severe | Mild | Severe | Severe | Mild |
| Portal-based fibrosis | Cirrhosis | Absent | Periportal | Periportal | Periportal |
| Patient No. | PFIC type | Gene | Mutation | Mutation type | BSEP immunostaining |
|---|---|---|---|---|---|
| 1 | 2 | ABCB11 on 2q24 | Absent | NA | Total loss |
| 2 | 2 | ABCB11 on 2q24 | c.1907A > G (p.Glu636Gly) (p.E636G) | Missense, heterozygous | Focal loss |
| c.2594C > T (p.Ala865Val) (p.A865V)a | Missense, heterozygous | ||||
| 3 | 2 | ABCB11 on 2q24 | Absent | NA | Total loss |
| 4b | 2 | ABCB11 on 2q24 | c.1416T > A (p.Tyr472*) (p.Y472*) | Nonsense, homozygous | Focal loss |
| 5 | 1 | ATP8B1 on 18q21 | c.1753G > T (p.Glu585*) (p.E585*) | Nonsense, heterozygous | Intact |
| c.2246T > C (p.Leu749Pro) (p.L749P)a | Missense, heterozygous |
- 1. Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis 2009; 4: 1.ArticlePubMedPMCPDF
- 2. Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 2012; 36 Suppl 1: S26-35. ArticlePubMed
- 3. Clayton RJ, Iber FL, Ruebner BH, McKusick VA. Byler disease. Fatal familial intrahepatic cholestasis in an Amish kindred. Am J Dis Child 1969; 117: 112-24. ArticlePubMed
- 4. Bull LN, van Eijk MJ, Pawlikowska L, et al. A gene encoding a Ptype ATPase mutated in two forms of hereditary cholestasis. Nat Genet 1998; 18: 219-24. ArticlePubMedPDF
- 5. Strautnieks SS, Bull LN, Knisely AS, et al. A gene encoding a liverspecific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 1998; 20: 233-8. ArticlePubMedPDF
- 6. van Mil SW, Klomp LW, Bull LN, Houwen RH. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin Liver Dis 2001; 21: 535-44. ArticlePubMedPDF
- 7. Stapelbroek JM, van Erpecum KJ, Klomp LW, Houwen RH. Liver disease associated with canalicular transport defects: current and future therapies. J Hepatol 2010; 52: 258-71. ArticlePubMed
- 8. Park YN, Kim HG, Chon CY, et al. Histological grading and staging of chronic hepatitis standardized guideline proposed by the Korean Study Group for the Pathology of Digestive Diseases. Korean J Pathol 1999; 33: 337-46.
- 9. Yu E, Korean Study Group for the Pathology of Digestive Diseases. Histologic grading and staging of chronic hepatitis: on the basis of standardized guideline proposed by the Korean Study Group for the Pathology of Digestive Diseases. Taehan Kan Hakhoe Chi 2003; 9: 42-6. PubMed
- 10. Nemesánszky E, Lott JA. Gamma-glutamyltransferase and its isoenzymes: progress and problems. Clin Chem 1985; 31: 797-803. ArticlePubMedPDF
- 11. Oude Elferink RP, Paulusma CC, Groen AK. Hepatocanalicular transport defects: pathophysiologic mechanisms of rare diseases. Gastroenterology 2006; 130: 908-25. ArticlePubMed
- 12. Evason K, Bove KE, Finegold MJ, et al. Morphologic findings in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with genetic and immunohistochemical studies. Am J Surg Pathol 2011; 35: 687-96. PubMedPMC
- 13. Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr 2008; 46: 241-52. ArticlePubMed
- 14. Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis 2011; 31: 3-10. ArticlePubMedPDF
- 15. Hori T, Nguyen JH, Uemoto S. Progressive familial intrahepatic cholestasis. Hepatobiliary Pancreat Dis Int 2010; 9: 570-8. PubMed
- 16. Bull LN, Carlton VE, Stricker NL, et al. Genetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC-1] and Byler syndrome): evidence for heterogeneity. Hepatology 1997; 26: 155-64. ArticlePubMed
- 17. Gonzales E, Spraul A, Jacquemin E. Clinical utility gene card for: progressive familial intrahepatic cholestasis type 1. Eur J Hum Genet 2013 Aug 28 [Epub]. https://doi.org/10.1038/ejhg.2013.186. Article
- 18. Gonzales E, Spraul A, Jacquemin E. Clinical utility gene card for: progressive familial intrahepatic cholestasis type 2. Eur J Hum Genet 2013 Aug 28 [Epub]. https://doi.org/10.1038/ejhg.2013.187. Article
- 19. Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology 2004; 40: 27-38. ArticlePubMed
- 20. Liu LY, Wang XH, Wang ZL, Zhu QR, Wang JS. Characterization of ATP8B1 gene mutations and a hot-linked mutation found in Chinese children with progressive intrahepatic cholestasis and low GGT. J Pediatr Gastroenterol Nutr 2010; 50: 179-83. ArticlePubMed
- 21. Jacquemin E, Hermans D, Myara A, et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology 1997; 25: 519-23. ArticlePubMed
- 22. Amer S, Hajira A. A comprehensive review of progressive familial intrahepatic cholestasis (PFIC): genetic disorders of hepatocanalicular transporters. Gastroenterology Res 2014; 7: 39-43. ArticlePubMedPMC
- 23. Bustorff-Silva J, Sbraggia Neto L, Olímpio H, et al. Partial internal biliary diversion through a cholecystojejunocolonic anastomosis: a novel surgical approach for patients with progressive familial intrahepatic cholestasis: a preliminary report. J Pediatr Surg 2007; 42: 1337-40. ArticlePubMed
- 24. Aydogdu S, Cakir M, Arikan C, et al. Liver transplantation for progressive familial intrahepatic cholestasis: clinical and histopathological findings, outcome and impact on growth. Pediatr Transplant 2007; 11: 634-40. ArticlePubMed
- 25. Hori T, Egawa H, Takada Y, et al. Progressive familial intrahepatic cholestasis: a single-center experience of living-donor liver transplantation during two decades in Japan. Clin Transplant 2011; 25: 776-85. ArticlePubMed
- 26. Englert C, Grabhorn E, Richter A, Rogiers X, Burdelski M, Ganschow R. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation 2007; 84: 1361-3. ArticlePubMed
- 27. Lykavieris P, van Mil S, Cresteil D, et al. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol 2003; 39: 447-52. ArticlePubMed
- 28. Miyagawa-Hayashino A, Egawa H, Yorifuji T, et al. Allograft steatohepatitis in progressive familial intrahepatic cholestasis type 1 after living donor liver transplantation. Liver Transpl 2009; 15: 610-8. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Exploring the interplay of Emotional intelligence and stress
Ana-Lucia Blendea, Ioan Gotcă , Teodora-ELena Huțanu , Alin Ciobîcă , Daniela Dumitriu
Bulletin of Integrative Psychiatry.2024; 101(2): 45. CrossRef - Hepatocellular carcinoma associated with progressive intrahepatic familial cholestasis type 2: a case report
João Miguel Pimentel, Susana Nobre, Rui Caetano Oliveira, Ricardo Martins, Maria Augusta Cipriano
Clinical Transplantation and Research.2024; 38(3): 241. CrossRef - Progressive Familial Intrahepatic Cholestasis: A Descriptive Study in a Tertiary Care Center
Fahad I. Alsohaibani, Musthafa C. Peedikayil, Abdulaziz F. Alfadley, Mohamed K. Aboueissa, Faisal A. Abaalkhail, Saleh A. Alqahtani, Dirk Uhlmann
International Journal of Hepatology.2023; 2023: 1. CrossRef - Next-generation sequencing panel test results in pediatric patients with progressive familial intrahepatic cholestasis: a single-center experience
Ali TOPAK
The European Research Journal.2023; 9(6): 1438. CrossRef - Progressive Familial Intrahepatic Cholestasis: A Study in Children From a Liver Transplant Center in India
Sagar Mehta, Karunesh Kumar, Ravi Bhardwaj, Smita Malhotra, Neerav Goyal, Anupam Sibal
Journal of Clinical and Experimental Hepatology.2022; 12(2): 454. CrossRef - Liver transplantation in pediatric patients with progressive familial intrahepatic cholestasis: Single center experience of seven cases
Jung-Man Namgoong, Shin Hwang, Hyunhee Kwon, Suhyeon Ha, Kyung Mo Kim, Seak Hee Oh, Seung-Mo Hong
Annals of Hepato-Biliary-Pancreatic Surgery.2022; 26(1): 69. CrossRef - Liver Transplantation for Pediatric Hepatocellular Carcinoma: A Systematic Review
Christos D. Kakos, Ioannis A. Ziogas, Charikleia D. Demiri, Stepan M. Esagian, Konstantinos P. Economopoulos, Dimitrios Moris, Georgios Tsoulfas, Sophoclis P. Alexopoulos
Cancers.2022; 14(5): 1294. CrossRef - Morphology of transplanted liver in recurrent progressive familial intrahepatic cholestasis type 2
I. M. Iljinsky, N. P. Mozheiko, O. M. Tsirulnikova
Russian Journal of Transplantology and Artificial Organs.2021; 22(4): 192. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
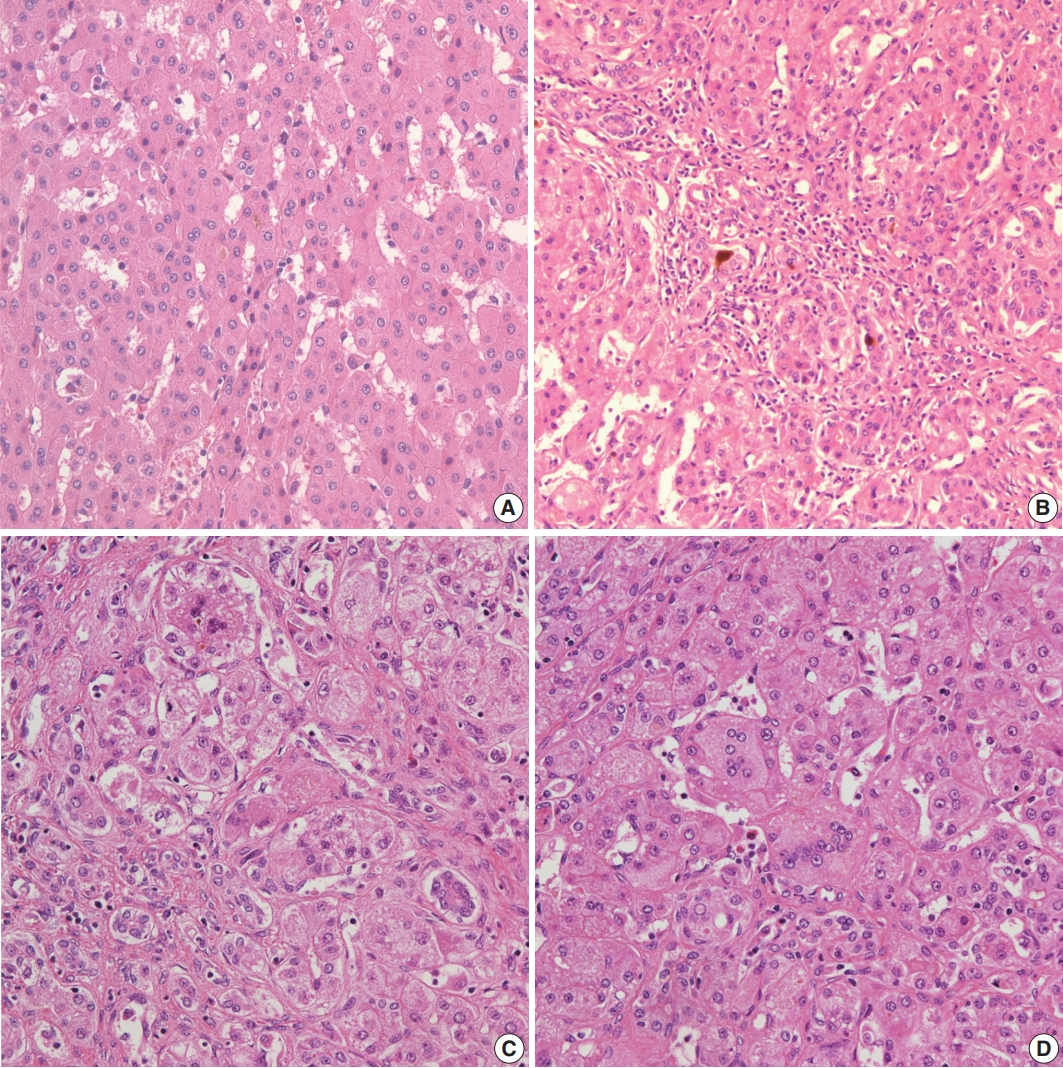
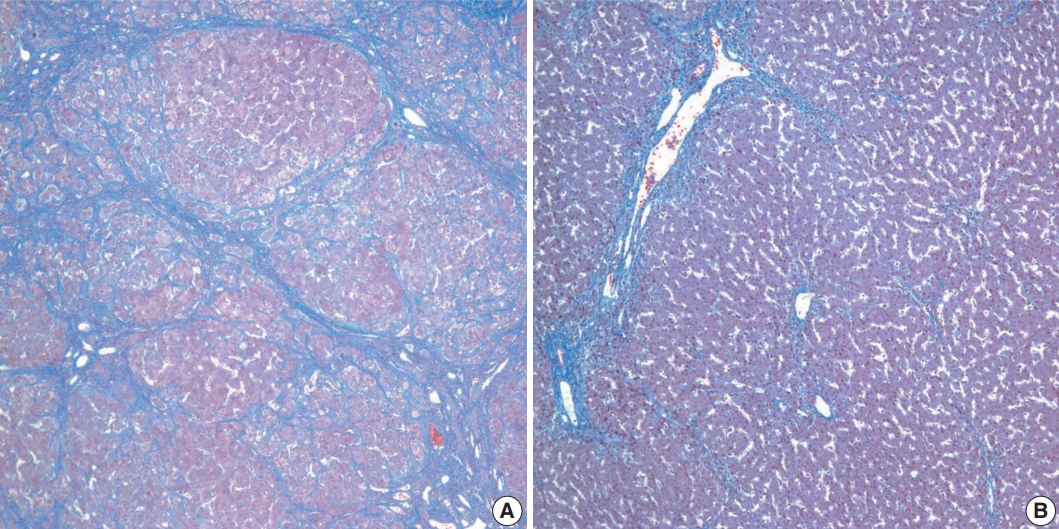
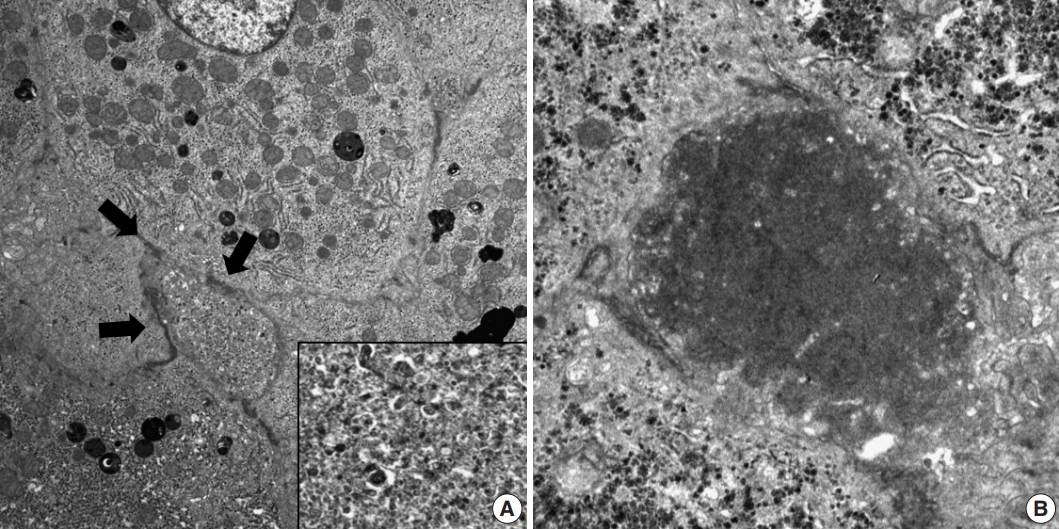
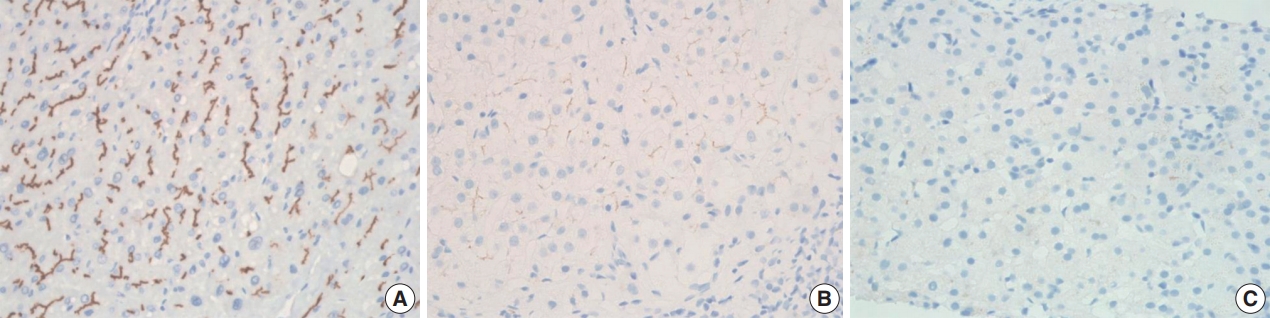




Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
| Patient No. | PFIC type | Sex/Age at onset | Initial symptom | AST (IU/L) | ALT (IU/L) | GGT (mg/dL) | ALP (IU/L) | TB (mg/dL) | DB (mg/dL) | Concomitant tumor | Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | F/Birth | Jaundice | 282 | 152 | 32 | 356 | 24.4 | 21.0 | HCC, infantile hemangioma | LT |
| 2 | 2 | M/45 days | Jaundice | 72 | 65 | 15 | 594 | 10 | 5.6 | Absent | UDCA |
| 3 | 2 | F/Birth | Jaundice | 43 | 20 | 57 | 576 | 10.6 | 7.6 | Absent | LT |
| 4 |
2 | F/Birth | Jaundice | 468 | 553 | 30 | 402 | 20.0 | 15.0 | Absent | LT |
| 5 | 1 | F/Birth | Jaundice | 69 | 42 | 16 | 824 | 19.6 | 19.0 | Absent | LT |
| Pathologic parameter | Case 1 (PFIC2) | Case 2 (PFIC2) | Case 3 (PFIC2) | Case 4 |
Case 5 (PFIC1) |
|---|---|---|---|---|---|
| Specimen type | Biopsy (× 1) | Biopsy (×1) | Biopsy (× 1) | Posttansplant Bx (×3) | Biopsy (× 1) |
| Explanted liver (× 1) | Explanted liver (× 1) | Explanted liver (× 1) | |||
| Posttansplant Bx (× 1) | Posttransplant Bx (4) | ||||
| Bile plugs | Canalicular/Periportal/Cholangiolar | Canalicular | Canalicular/Periportal | Canalicular/Periportal | Canalicular |
| Lobular activity | Absent | Absent | Severe | Mild | Absent |
| Giant cell change | Present | Absent | Present | Present | Absent |
| Ballooning change | Present | Absent | Present | Present | Absent |
| Small cell change | Absent | Absent | Absent | Absent | Present |
| Portal activity | Mild | Absent | Mild | Mild | Mild |
| Ductular reaction | Present | Present | Present | Present | Present |
| Lobular-based fibrosis | Severe | Mild | Severe | Severe | Mild |
| Portal-based fibrosis | Cirrhosis | Absent | Periportal | Periportal | Periportal |
| Patient No. | PFIC type | Gene | Mutation | Mutation type | BSEP immunostaining |
|---|---|---|---|---|---|
| 1 | 2 | ABCB11 on 2q24 | Absent | NA | Total loss |
| 2 | 2 | ABCB11 on 2q24 | c.1907A > G (p.Glu636Gly) (p.E636G) | Missense, heterozygous | Focal loss |
| c.2594C > T (p.Ala865Val) (p.A865V) |
Missense, heterozygous | ||||
| 3 | 2 | ABCB11 on 2q24 | Absent | NA | Total loss |
| 4 |
2 | ABCB11 on 2q24 | c.1416T > A (p.Tyr472*) (p.Y472*) | Nonsense, homozygous | Focal loss |
| 5 | 1 | ATP8B1 on 18q21 | c.1753G > T (p.Glu585*) (p.E585*) | Nonsense, heterozygous | Intact |
| c.2246T > C (p.Leu749Pro) (p.L749P) |
Missense, heterozygous |
PFIC, progressive familiar intrahepatic cholestasis; AST, asparate aminotransferase; ALT, alanine aminotransferase; GGT, gamma-glutamyl transpeptidase; ALP, alkaline phosphatase; TB, total bilirubin; DB, direct bilirubin; F, female; HCC, hepatocellular carcinoma; LT, liver transplantation; M, male; UDCA, urodeoxycholic acid. Arabian.
PFIC, progressive familiar intrahepatic cholestasis; Bx, biopsy. Arabian.
BSEP, bile salt export pump; PFIC, progressive familial intrahepatic cholestasis; NA, not available. Possible mutation; Arabian.

