 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 58(1); 2024 > Article
-
Review
Diagnosis of interstitial lung diseases: from Averill A. Liebow to artificial intelligence -
Eunhee S. Yi,1
 , Paul Wawryko2, Jay H. Ryu3
, Paul Wawryko2, Jay H. Ryu3 -
Journal of Pathology and Translational Medicine 2024;58(1):1-11.
DOI: https://doi.org/10.4132/jptm.2023.11.17
Published online: January 10, 2024
1Division of Anatomic Pathology, Mayo Clinic Rochester, Rochester, MN, USA
2Division of Anatomic Pathology, Mayo Clinic Arizona, Arizona, FL, USA
3Division of Pulmonary and Critical Medicine, Mayo Clinic Rochester, Rochester, MN, USA
- Corresponding Author: Eunhee S. Yi, MD, Division of Anatomic Pathology, Mayo Clinic Rochester, 200 First Street SW, Rochester, MN 55905, USA Tel: +1-507-284-2656, Fax: +1-507-266-3771, E-mail: Yi.joanne@mayo.edu
© 2024 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Histopathologic criteria of usual interstitial pneumonia (UIP)/idiopathic pulmonary fibrosis (IPF) were defined over the years and endorsed by leading organizations decades after Dr. Averill A. Liebow first coined the term UIP in the 1960s as a distinct pathologic pattern of fibrotic interstitial lung disease. Novel technology and recent research on interstitial lung diseases with genetic component shed light on molecular pathogenesis of UIP/IPF. Two antifibrotic agents introduced in the mid-2010s opened a new era of therapeutic approaches to UIP/IPF, albeit contentious issues regarding their efficacy, side effects, and costs. Recently, the concept of progressive pulmonary fibrosis was introduced to acknowledge additional types of progressive fibrosing interstitial lung diseases with the clinical and pathologic phenotypes comparable to those of UIP/IPF. Likewise, some authors have proposed a paradigm shift by considering UIP as a stand-alone diagnostic entity to encompass other fibrosing interstitial lung diseases that manifest a relentless progression as in IPF. These trends signal a pendulum moving toward the tendency of lumping diagnoses, which poses a risk of obscuring potentially important information crucial to both clinical and research purposes. Recent advances in whole slide imaging for digital pathology and artificial intelligence technology could offer an unprecedented opportunity to enhance histopathologic evaluation of interstitial lung diseases. However, current clinical practice trends of moving away from surgical lung biopsies in interstitial lung disease patients may become a limiting factor in this endeavor as it would be difficult to build a large histopathologic database with correlative clinical data required for artificial intelligence models.
- Usual interstitial pneumonia
- The histologic classification of interstitial pneumonias evolved in a time when surgical lung biopsies were infrequently performed owing to significant morbidity associated with the surgical techniques of the day. As a result, histologic findings did not inform clinical decision-making in most patients with interstitial pneumonia. Clinicians were left to diagnose and manage patients with ILD primarily based on clinical and radiologic features; clinical concepts did not evolve in concert with the histologic classification. In this background, idiopathic pulmonary fibrosis (IPF) originated as a clinical diagnosis without established histopathologic correlates.
- The original histologic description of UIP contained a wide spectrum of findings including acute lung injury in the form of what today would be considered diffuse alveolar damage, as well as chronic fibrosis and end-stage lung [2]. Although the histologic findings considered diagnostic of UIP were refined over time, established diagnostic criteria were still lacking into the 1990s with many investigators simply requiring the presence of inflammation and fibrosis in various proportions as diagnostic of UIP [3]. Many also considered even DIP to represent an early “cellular stage” of UIP [2].
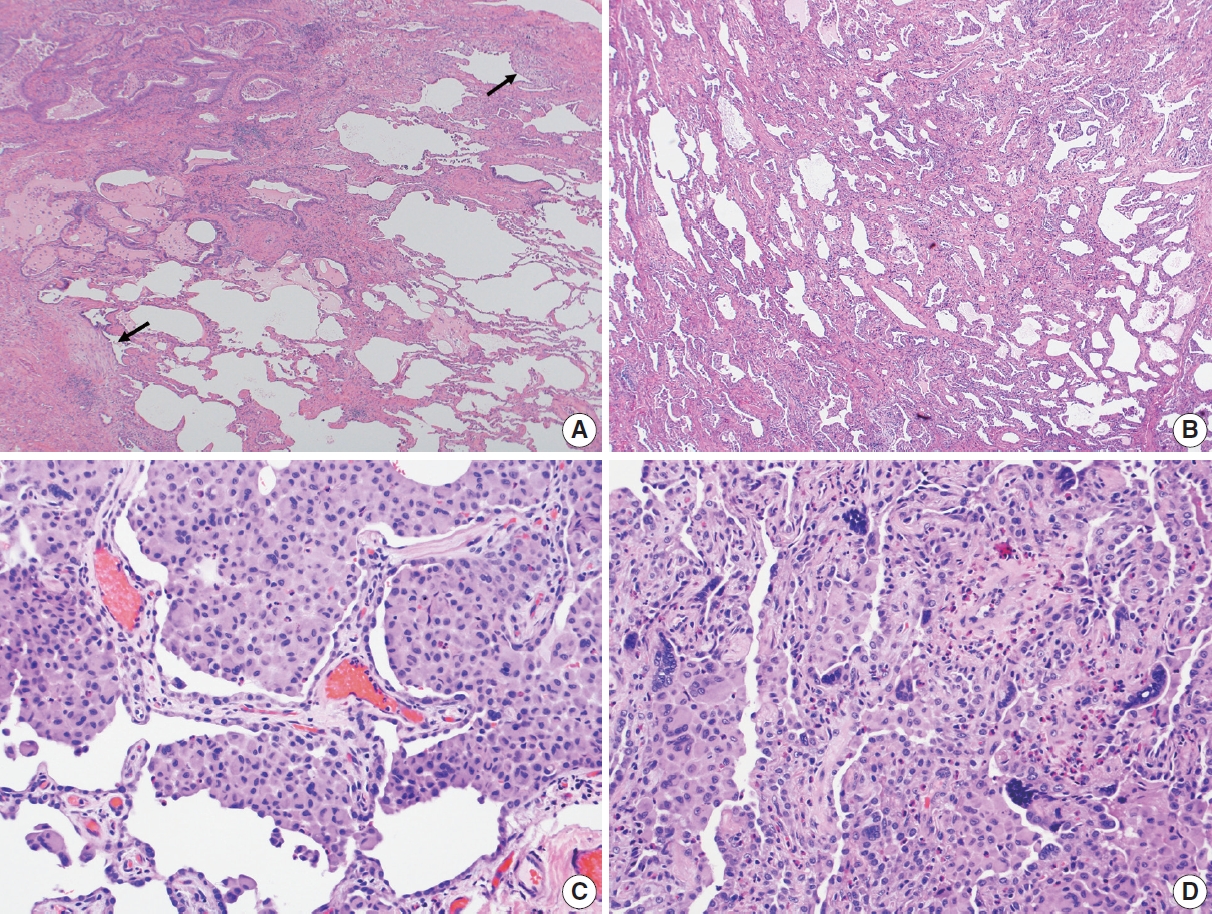
- In 1998, Katzenstein and Myers [2] described the crucial histologic features that were reflected in the ATS/ERS international consensus statement and established as the current histopathologic criteria of UIP [2,4]. The most recent 2022 ATS/ERS/JRS/ALAT clinical practice guideline used the same criteria including (1) patchy, dense fibrosis with architectural distortion causing destructive scarring and/or honeycombing, (2) predominantly subpleural and/or paraseptal distribution of fibrosis, (3) temporal heterogeneity of fibrosis characterized by fibroblast foci over scarred lung tissue, and (4) absence of features suggesting an alternate diagnosis (including prominent airway-centered changes, significant degree of OP, granulomas, hyaline membranes, dense lymphoid infiltrates, and marked chronic pleuritis, etc.) [4]. Different levels of confidence such as diagnostic of UIP (meeting all criteria) and probable UIP (having only some of these features) were proposed for pathologic diagnosis categories as in radiologic counterparts. This type of categorization may be useful in the clinical trial setting to recruit patients although it has not been widely applied in routine pathology practice.
- In the 2013 ATS/ERS statement, the major IIPs were subdivided into three categories: chronic fibrosing interstitial pneumonias (IPF and NSIP), smoking-related interstitial pneumonias (respiratory bronchiolitis-ILD and DIP), and acute/subacute interstitial pneumonias (cryptogenic OP and AIP) [5]. The classification also includes rare IIPs (idiopathic LIP and idiopathic pleuroparenchymal fibroelastosis) and unclassifiable IIP. Each of the IIPs has an associated radiologic and/or pathologic-morphologic pattern (e.g., UIP for IPF) and is considered a “clinical-radiologic-pathologic diagnosis,” emphasizing the multidisciplinary approach to diagnosis.
- The role of surgical lung biopsy in the diagnosis of IPF has changed over time. With the original ATS statement on IPF, surgical lung biopsy was recommended in most patients to establish the diagnosis, especially in those with atypical clinical or radiologic features for IPF [4]. The 2002 ATS/ERS IIP guidelines stated that biopsy was required for a confident diagnosis of UIP/IPF, but also noted that in more than 50% of cases of suspected IPF, the presence of typical clinical and HRCT features of UIP was sufficiently characteristic to allow a confident diagnosis without the need for surgical lung biopsy [4]. In the 2011 ATS statement of IPF, radiologic and pathologic criteria were developed to establish confidence categories for UIP (i.e. UIP pattern, probable UIP pattern, possible UIP pattern); thus, surgical lung biopsy was no longer required for histopathologic confirmation in patients with HRCT showing typical UIP pattern [6]. The only change regarding sampling since that time was the adoption of transbronchial lung cryobiopsy as an acceptable alternative to surgical lung biopsy in patients with ILD of undetermined type based on evidence that there was often diagnostic agreement between transbronchial cryobiopsy and surgical lung biopsy samples [7].
- Patients with IPF undergo a progressive decline in pulmonary function with eventual death from either respiratory failure or a complicating comorbidity, with a median survival of 2 to 3 years from the time of diagnosis [6]. Acute exacerbation may occur with histologic manifestions of diffuse alveolar damage or OP, though less common [10]. Based on the hypothesis that inflammation might be the underlying cause of lung injury and fibrosis, corticosteroids and other immunosuppressive agents have been tried without success. No effective pharmacologic therapy for IPF was available until the early 2010s, when two promising antifibrotic agents emerged. Randomized trials showed in disease progression and rate of forced vital capacity decline in IPF patients treated with nintedanib, a tyrosine kinase inhibitor, or pirfenidone, an inhibitor of transforming growth factor β–associated collagen synthesis [11-14]. These two medications were recommended for use in IPF patients as of the 2015 treatment guidelines [15]. Data from more recent clinical trials led to an expansion of antifibrotic therapy to other types of fibrosing ILD if they show evidence of clinical, physiologic, or radiologic progression [4].
- As such, diagnostic paradigm seems to have shifted to categorize the ILDs into two groups: cases to be treated vs. not to be treated with antifibrotics, a transition from the previous paradigm: to give or not to give corticosteroids (or other immunosuppressants). The recent proposal advocating UIP as a stand-alone diagnostic entity regardless of underlying causes is at least partly based on this therapeutic paradigm. However, such a lumping of various fibrosing ILDs might not be entirely justified, given the limited efficacy, significant side effects, and high cost of currently available antifibrotics as well as the risk of losing potentially useful diagnostic granularity. This proposal is reviewed in the section below. Similarly, the recent concept of progressive pulmonary fibrosis (PPF) published in the 2022 ATS guidelines would be in keeping with such a trend of lumping ILD diagnoses (see Concept of PPF: green light for lumping fibrosing ILD diagnoses, as exactly shown in the manuscript).
- UIP is generally regarded as the correlate of IPF. Accordingly, the cases with histopathologic features of other diseases such as fibrotic HP and CTD-related ILD are considered not consistent with UIP [16]. It is not uncommon, however, to have some minor changes associated with HP, CTD-related ILD, or other diseases (e.g., a few poorly formed granulomas, interstitial chronic inflammatory infiltrates, lymphoid hyperplasia, and airway-centered fibrosis, etc.) in the cases otherwise acceptable for UIP. Pathologists are often in a difficult position to determine whether the presence of these minor changes would disqualify the diagnosis of UIP or not. There have been widely differing opinions and approaches in this regard even among expert pulmonary pathologists.
- In this backdrop of diagnostic conundrum, a bold concept was proposed to consider UIP as a stand-alone diagnostic entity, encompassing not only IPF but also other secondary processes, based on the presence of radiologic or histopathologic features of UIP [17]. It was argued that UIP pattern of fibrosis in other ILDs (especially fibrotic HP and CTD-associated ILD) as well as in IPF is associated with unfavorable clinical outcomes [18]. In addition, acute exacerbation may occur in rheumatoid arthritis–associated ILD and fibrotic HP with similarly dismal outcome to that seen in IPF [19,20]. The significant similarities between IPF and other secondary conditions with UIP features in clinical behavior, pathogenic pathways, and the efficacy of anti-fibrotic therapy were cited as the justification for a lumping approach in this proposal. A radiologic study reported that the presence of honeycomb change alone predicted an IPF-like mortality in patients with other conditions (fibrotic HP, CTD-ILD, and unclassifiable ILD), which could support this notion [21].
- Concept of PPF: green light for lumping fibrosing ILD diagnoses
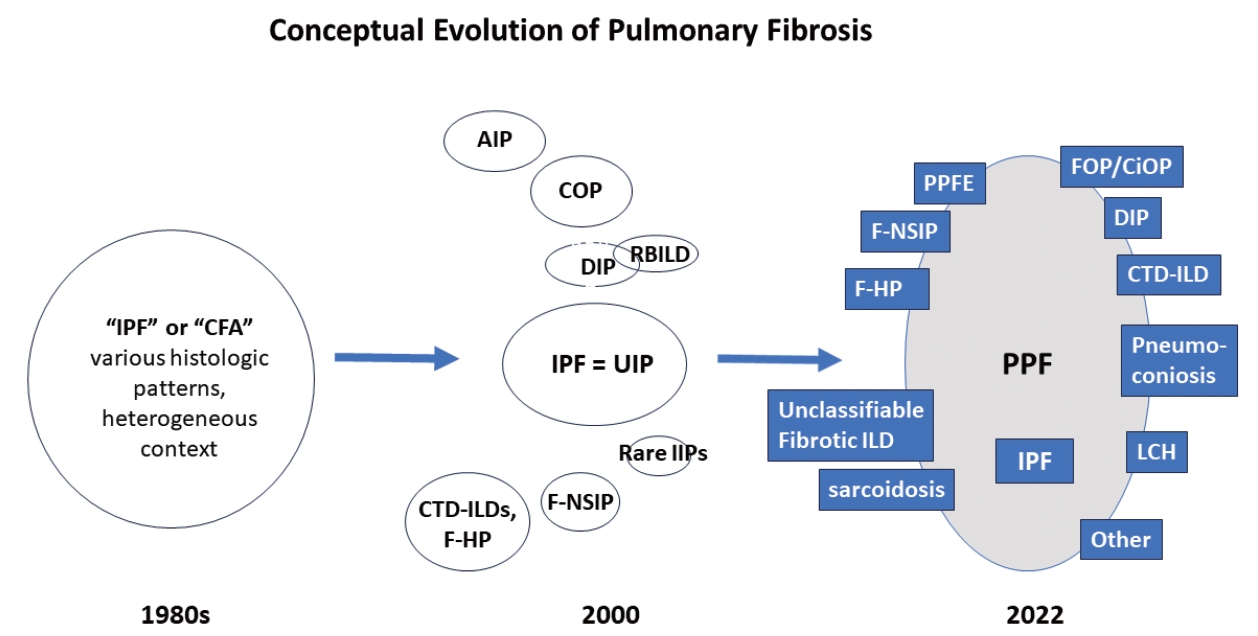
- There are many types of idiopathic and secondary fibrosing ILDs, in which IPF is considered as the prototype [22]. Non-IPF fibrosing ILDs include other IIPs (e.g., fibrotic NSIP, DIP, and AIP, etc.), autoimmune ILDs (e.g. rheumatoid arthritis-associated ILD), exposure-related ILDs (e.g. HP, occupational exposures, medications), pulmonary Langerhans cell histiocytosis and sarcoidosis, among others [7]. Recently, fibrosing ILDs have been studied as a group, given their clinical, radiological, and histopathological overlap. In this approach, a subset of non-IPF fibrosing ILDs was found to show a very similar clinical phenotype to that of IPF, characterized by progressive worsening of respiratory symptoms, declining lung function, developing acute exacerbation, and resistance to conventional therapy, ultimately resulting in high mortality [23-26]. In 2017, a term “progressive fibrosing ILDs” was first coined to this group of cases but an alternative term “progressive pulmonary fibrosis” (PPF) was endorsed in the 2022 ATS/ERS/JRS/ALAT clinical practice guideline [7,26]. In this 2022 statement, PPF was officially defined as an ILD of known or unknown etiology, other than IPF, with radiologic evidence of pulmonary fibrosis as well as meeting the set of specific clinical/radiological criteria including worsening respiratory symptoms and a certain degree of progression in pulmonary function abnormalities and/or radiological evidence of disease progression, occurring within the past year [7]. In this guideline, PPF is clearly stated as an entity of prognostic significance but not a specific diagnosis per se. The conceptual evolution to PPF over time is illustrated in Fig. 3.
- Clinical trials have evaluated the efficacy of antifibrotics approved for IPF in the non-IPF patients who underwent progressive fibrosis, regardless of the type of underlying ILDs. Nintedanib therapy in the INBUILD trial resulted in a significant reduction in the annual decline of FVC in patients with PPF [27]. Pirfenidone therapy in two randomized control trials also showed a reduced FVC decline in some PPF patients [28,29]. Based on the results of these trials, the 2022 ATS clinical practice guidelines gave a conditional recommendation for nintedanib for the treatment of PPF in patients who have failed standard management, but not for pirfenidone given the lack of sufficient evidence [7].
- This novel approach to treat ILD based on the disease behavior (i.e., PPF) without consideration of the specific underlying diagnosis will have a broader, potentially deleterious, influence in many aspects of ILD. The ATS guideline on PPF explicitly stated that this new concept of PPF should not discourage clinicians from their rigorous effort to identify the underlying type of ILD before antifibrotic therapy. In reality, however, it is most likely that the incentive of establishing a definitive diagnosis of the underlying ILD would be markedly reduced if not completely vanished. A few studies attempted to analyze the clinical trials data based on specific ILD subgroups within PPF but did not have enough power to provide evidence of benefit in different subgroups [30,31]. The criteria of PPF do not include pathologic findings, but it may be helpful to evaluate any histopathologic findings that correlate with progressive fibrosis. While UIP histopathologic pattern is an obvious leading candidate given its well-known compelling evidence, characterization of other histologic features that could predict the PPF phenotype has yet to be established [17].
- Familial pulmonary fibrosis: a clue to decode molecular pathogenesis of fibrosing ILD
- Familial cases of ILDs were reported in 1907, which suggested a possibility of genetic factors in ILDs [32]. The concept of familial IPF was first introduced in the 2000 ATS statement with the following definition: at least two members of a primary biological family (parent, child, sibling) with clinical features of IPF and histologic confirmation [3]. Though not explicitly recommended in later ATS statements, the histology requirement in the criteria was eventually dropped in practice, which is in keeping with the trends of bypassing the biopsies and making IPF diagnosis based on HRCT at least in more classic cases of IPF on clinical ground. Familial clustering of non-IPF fibrosing ILDs have also been observed. Other names have been used for this group of diseases, including familial/inherited interstitial lung disease, familial interstitial pneumonia, familial pulmonary fibrosis, and familial IIP, if limited to idiopathic cases. The plethora of terminology has clouded the literature and hampered clearer communication. In this review, the term familial pulmonary fibrosis (FPF) will be used that encompasses all types of pulmonary fibrosis including IPF.
- The frequency of FPF may be as high as 20% of patients with pulmonary fibrosis according to the previous studies reported in the literature [33-38]. Among the subtypes of fibrotic ILD, IPF most frequently had a family history of pulmonary fibrosis (20%– 25%), followed by chronic HP (14%–17%), and CTD-related ILD (3%–8%) [38-41]. Of note, relatives in the same family may show different subtypes of fibrotic ILD [42,43]. Radiographic screening studies revealed some radiological abnormalities in 15 to 31% of the asymptomatic relatives of patients with known pulmonary fibrosis, suggesting even higher prevalence of FPF than suspected [44-46].
- The remarkable advances in molecular technology led to several large-scale, population-based studies on pulmonary fibrosis to identify genetic risk variants that could be associated with crucial pathogenetic mechanisms in pulmonary fibrosis. Two broad categories of the variants associated with pulmonary fibrosis have been recognized based on their frequency in the population: common and rare genetic variants. Common genetic variants typically represent single nucleotide polymorphisms (SNPs) and confer a smaller effect than rare variants as the impact on disease risk tends to be inversely proportional to the frequency of a variant within the population [47]. It has been well accepted that SNPs may contribute to overall risk but not entirely sufficient to cause disease on their own. On the other hand, rare variants often demonstrate cosegregation in FPF kindreds, suggesting a causal relationship.
- Linkage analysis and genome-wide association studies have identified numerous common genetic variants associated with IPF. Gain-of-function promoter variant rs35705950 in the promoter region of the MUC5B gene is most widely recognized in association with pulmonary fibrosis. A study reported that this genetic variant is identified in 34% of the subjects with familial IIP, 38% of those with sporadic IPF, and 9% of the control subjects [48].
- Rare genetic variants in pulmonary fibrosis were mainly implicated in dysfunctional surfactant metabolism and telomere maintenance. Genes involved in abnormal surfactant metabolism include SFTPC, SFTPA1/2, and ABCA3 [40]. The pattern of inheritance is autosomal dominant for SFTPC and SFTPA1/2 and autosomal recessive for ABCA3 [49]. Disease onset has a wide range from infancy to late adulthood in families with a rare surfactant-related gene variant; histologic and radiologic features of ILD are also diverse with the features of UIP, NSIP, and DIP [50-52]. No extrapulmonary manifestations are present in these patients as surfactant production is limited to the lung. The rare genetic variants associated with deranged telomere maintenance account for approximately 25% of FPF kindreds and involve TERT, TERC, and numerous other genes [53,54]. Unlike the gene variants associated with surfactant dysfunction, these variants of telomere-related genes often cause extrapulmonary systemic manifestations (known as short telomere syndrome or other related terms) [55]. Dyskeratosis congenita (DC) is one of the best known telomeropathy due to homozygous telomere-related gene mutations resulting in extreme telomere shortening. DC causes pulmonary fibrosis (20%) as well as life-threatening bone marrow failure (80%) at pediatric age or in very early adulthood. DC often shows various cutaneous or mucosal manifestations including nail dystrophy, abnormal skin pigmentation, and oral leukoplakia [55,56].
- The patients with heterozygous telemere–related gene mutations also often develop pulmonary fibrosis including IPF (50%), fibrotic HP (7%–12%) connective tissue disease–associated ILD (2%–3%), or other IIPs (14%–18%) with similar extrapulmonary manifestations of DC but at an older age than in DC [43,57,58]. Interestingly, about 10% of adult-onset sporadic IPF, chronic HP, and rheumatoid arthritis-related ILD patients have rare telomererelated gene variants [59-62]. Short telomere length is often seen in FPF cases and a frequent finding in sporadic cases of pulmonary fibrosis as compared to control subjects, suggesting a possibility that short telomere length might be a potential cause of pulmonary fibrosis [63].
- Despite a remarkable progress in identifying key genetic features associated with FPF (and some sporadic fibrosing ILD cases), characterization of the matching pathologic features in FPF seemed to have lagged behind. Steele et al published a study in 2005 with histopathologic assessment in some of their cases from families with IIPs and found significant heterogeneity within a given family, showing more than one histopathologic subtype of IIP in 45% of family pedigrees [42]. A comprehensive histopathologic study by Leslie et al compiled the findings to differentiate familial and sporadic IIP cases in an effort to characterize the features of familial IIP, primarily focusing on familial IPF [64]. Although most of their patients had some histopathologic features associated with UIP, up to 60% of them did not qualify as UIP, mainly due to lack of the temporal heterogeneity of fibrosis, one of the most important histologic criteria for UIP [64]. Most of these cases ended up in the unclassifiable fibrotic ILD category [64]. Prevailing notion in the past has been that familial and sporadic IPF cases do not have distinguishable clinical or histological features other than earlier onset in familial IPF [6]. However, this 2012 study by Leslie et al. [64] suggested that there may be some histopathologic features differentiating sporadic and familial fibrotic lung diseases. Based on this observation, they concluded that unclassifiable pattern of lung fibrosis should raise a possibility of FPF or familial IPF [64]. They also suggested that less than classic histopathologic features of UIP could still be compatible with familial IPF in an appropriate clinical context [64].
- AI in diagnosis of interstitial lung diseases
- The application of AI tools on radiologic images for characterization of indeterminate lung nodules, fibrotic lung diseases, and lung cancer risk stratification has been well studied and documented in the literature. As compared to radiology arena, digital pathology (DP) by WSI started more recently and most AI approach has been applied to cancers and biomarker arena of other organs (such as breast and prostate). In the lungs, AI tools have also been more widely applied to cancers than to ILDs, partly due to the current tendency of relying on high-resolution CT and bypassing surgical lung biopsies for diagnosis of ILDs, especially after the 2013 ATS/ERS guidelines.
- As there are many clinically as well as morphologically ambiguous cases of ILD, AI-assisted diagnosis may offer a crucial contribution by detecting some features that are not discernable by traditional diagnostic methods. Moreover, AI apporach may resolve some interobserver variability in diagnosing ILDs that has been well-recognized even among the expert pulmonary pathologists. It is inherently subjective to interpret the basic histopathologic parameters for ILD diagnosis (such as architectural distortion or scarring, honeycomb change, traction bronchiectasis, distribution of fibrosis, degree of inflammation, fibroblast foci, presence of granulomas, to name a few). Thus, more objective interpretation and quantitation of these parameters would be ideal by appropriate training and development of AI algorithm. Once AI-assisted diagnostic tool becomes widely available, it may mitigate the challenges encountered by many hospitals without direct access to experienced pathologists who are well versed in ILD diagnoses. Finally, some advanced AI technology (e.g. DL-based AI via unsupervised feature learning) might offer new insights into ILDs by elucidating previously unknown histopathologic features associated with progressive clinical behavior as in PPF cases. Such an approach, however, requires a large number of surgical lung biopsy cases with clinical annotation, which became scarce these days due to the changing practices as elaborated in the earlier sections. Multi-institutional collaboration might be needed to form a consortium for securing sufficient database to develop appropriate AI model.
- A complete coverage of AI tools in DP is beyond the scope of the present review and only a brief introduction of basic concepts in the AI approach will be provided, followed by summary of several original studies in the literature on predicting pathologic diagnosis and prognosis of ILDs, or evaluation of histopathologic biomarkers in IPF with AI technology (Table 1).
- AI is a broad discipline with multiple approaches to construct a model for a particular task. There are two common ways to extract feature representation for building an AI model: (1) DLbased unsupervised feature learning, (2) hand-crafted approach [65]. Convolutional neural network (CNN) is a type of DL approach suited for low-level tasks such as detecting, identifying, and classification of images. DL-based unsupervised feature learning is favored in low-level tasks such as cell and lung tumor detection and classification. This is useful since visual confirmation of the result is sufficient and does not require interpretation of selected features. In the hand-crafted approach, relevant features from data are manually selected. The domain knowledge is employed for features engineering, by close collaboration between pathologists and machine learning engineers to construct appropriate AI models. In contrast to DL-based unsupervised feature learning, hand-crafted based models offer some interpretability through incorporating the expertise of subject matter experts. Many high-level tasks such as prognosis or treatment response prediction which require a certain level of interpretability and hence hand-crafted, domain-inspired features may be favored by medical community to construct these models. Integration of DP in clinical workflow across major institutions poses a huge challenge, due to organizational structures, the cost of initial setup, requirements of advanced security systems in hospitals and demands for storage of big data. Currently, most existing AI tools have been validated on retrospective data, which does not represent the current, real-world scenarios. Validating the algorithms in randomized controlled trials and prospective studies will be a crucial step towards clinical adoption of these AI tools.
- Makela et al. [66] reported a study using AI to count fibroblast foci, a known prognostic factor in IPF, and other parameters to evaluate their prognostic significance. In this study, they trained a CNN to quantify fibroblast foci, interstitial mononuclear inflammation, and intra-alveolar macrophages to analyze the association of these parameters with survival [66]. Interstitial mononuclear inflammation and intra-alveolar macrophages were associated longer survival, while increased fibroblast foci were associated with poor prognosis.
- Uegami et al. [67] proposed an original method to develop DL models for extracting pathologically significant findings based on expert pathologist’s perspective with a small annotation effort. Their method named MIXTURE (huMan-In-the-loop eXplainable artificial intelligence Through the Use of REcurrent training) consisted three steps including (1) creating feature extractors for tiles from whole slide images using self-supervised learning, (2) clustering similar looking tiles based on output features, followed by integration of the pathologically synonymous clusters by pathologists, and (3) creation of DL models to classify tiles into pathological findings by using the integrated clusters as labeled data. They developed three models for different magnification. Their model predicted the diagnosis of UIP with a high accuracy, which was not possible to achieve without the step of integration of findings by pathologists. They proposed this model as the prototype for explainable AI that can collaborate with humans.
- Testa et al. [68] reported a study that performed an automated digital quantification of pulmonary fibrosis in human histopathology specimens. They conducted a pilot sutdy to analyze a small number of specimens from patients with Hermansky-Pudlak syndrome pulmonary fibrosis (n = 3) or IPF (n = 9) using digital images of serial lung sections stained with picrosirius red, alcian blue or anti-CD68 antibody. Dedicated software was used to automatically quantify fibrosis, collagen, and macrophage content. Automated fibrosis quantification based on parenchymal tissue density and fibrosis score measurements were compared to the pulmonary function test values or Ashcroft score, a numerical scale with grades from 0 to 8, of the amount of fibrotic tissue in histological samples devised by Ashcroft et al. [69]. A high correlation coefficient was found between some automated quantification measurements and lung function values in the sample groups. They concluded that computerized image analysis can offer accurate, reader-independent pulmonary fibrosis quantification in human histopathology samples for various parameters such as fibrosis, collagen content, and immunostained cells. This approach may enhance the available tools to quantify and study fibrotic ILDs.
SELECTED TOPICS
Evolution of histopathologic criteria for UIP
Recent trends of decreasing lung biopsy use
Antifibrotic therapy for patients with IPF and those with UIP due to underlying cause
Recent proposal of UIP as stand-alone entity
- Since Dr. Liebow introduced the diagnosis of UIP/IPF as a type of ILDs more than 50 years ago, histopathologic criteria have been refined and established. Antifibrotic therapy recently became available for UIP/IPF patients and brought modest improvement in its otherwise dismal clinical course. Partly based on the efficacy of antifibrotic therapy in other types of fibrosing ILDs, albeit limited, a proposal of UIP as a stand-alone entity was made to encompass all fibrosing ILDs with UIP pattern under the broad umbrella of UIP to be treated as UIP/IPF patients. Likewise, a concept of PPF that includes many types of fibrosing ILDs was endorsed as a prognostic entity (but not a specific diagnosis) by an international committee that also cited the similar therapeutic approach to that in UIP/IPF as the main reason for creating this category. Common and rare genetic variants identified in fibrosing ILD patients shed light in molecular pathways implicated in sporadic as well as familial cases of fibrosing ILDs. Whole slide imaging and AI might open the door to a new era provided accumulation of big histopathologic database and collective effort in this approach support the progress.
CONCLUSION
Ethics Statement
Not applicable.
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: ESY, JHR. Data curation: ESY, PW. Supervision: ESY. Writing—original draft: ESY, PW. Writing—review & editing: ESY, PW, JHR. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.
| Study | Purpose of study | Method | Country | No. of cases | Main finding |
|---|---|---|---|---|---|
| Makela et al. (2021) [66] | Quantitation of histologic parameters for survival analysis in IPF | Deep CNN | Finland | 71 IPF | Increased FF is associated with poor prognosis—interstitial mononuclear infiltrate & intra-alveolar macrophage with longer survival |
| Uegami et al. (2022) [67] | Prediction of UIP diagnosis | MIXTURE | Japan | 231 (UIP + non-UIP) cases; 715 WSI | A model approach to differentiate diseases by AI with collaboration with humans based on expert pathologists’ input |
| Testa et al. (2021) [68] | Pilot study to quantitate fibrosis in HPSPF & IPF | Automated quantitation of fibrosis with “dedicated software” | USA | 3 HPSPF & 9 IPF | Their automated image analysis offered accurate, reader-independent pulmonary fibrosis quantitation in HPSPF and IPF groups |
AI, artificial intelligence; CNN, convoluted neural network; IPF, idiopathic pulmonary fibrosis; FF, fibroblastic foci; MIXTURE, huMan-In-the loop eXpalainable artificial intelligence Through the Use of REcurrent training; UIP, usual interstitial pneumonia; WSI, whole slide imaging; HPSPF, Hermansky-Pudlak syndrome pulmonary fibrosis.
- 1. Liebow A, Carrington CB. The interstitial pneumonias. In: Simon M, Potchen EJ, LeMay M, eds. Frontiers of pulmonary radiology. New York: Grune and Stratton, 1969; 102-41.
- 2. Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 1998; 157: 1301-15. ArticlePubMed
- 3. American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med 2000; 161: 646-64. ArticlePubMed
- 4. American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 2002; 165: 277-304. ArticlePubMed
- 5. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188: 733-48. PubMedPMC
- 6. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788-824. PubMedPMC
- 7. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2022; 205: e18-47. PubMedPMC
- 8. Kim SY, Diggans J, Pankratz D, et al. Classification of usual interstitial pneumonia in patients with interstitial lung disease: assessment of a machine learning approach using high-dimensional transcriptional data. Lancet Respir Med 2015; 3: 473-82. ArticlePubMed
- 9. Raghu G, Flaherty KR, Lederer DJ, et al. Use of a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: a prospective validation study. Lancet Respir Med 2019; 7: 487-96. ArticlePubMed
- 10. Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2007; 176: 636-43. PubMed
- 11. Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2005; 171: 1040-7. ArticlePubMed
- 12. Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J 2010; 35: 821-9. ArticlePubMed
- 13. Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 2011; 365: 1079-87. ArticlePubMed
- 14. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071-82. PubMed
- 15. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis: an update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 2015; 192: e3-19. PubMed
- 16. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018; 198: e44-68. PubMed
- 17. Selman M, Pardo A, Wells AU. Usual interstitial pneumonia as a stand-alone diagnostic entity: the case for a paradigm shift? Lancet Respir Med 2023; 11: 188-96. ArticlePubMed
- 18. Hambly N, Farooqi MM, Dvorkin-Gheva A, et al. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur Respir J 2022; 60: 2102571.ArticlePubMed
- 19. Hozumi H, Nakamura Y, Johkoh T, et al. Acute exacerbation in rheumatoid arthritis-associated interstitial lung disease: a retrospective case control study. BMJ Open 2013; 3: e003132. ArticlePubMedPMC
- 20. Kang J, Kim YJ, Choe J, Chae EJ, Song JW. Acute exacerbation of fibrotic hypersensitivity pneumonitis: incidence and outcomes. Respir Res 2021; 22: 152.ArticlePubMedPMCPDF
- 21. Adegunsoye A, Oldham JM, Bellam SK, et al. Computed tomography honeycombing identifies a progressive fibrotic phenotype with increased mortality across diverse interstitial lung diseases. Ann Am Thorac Soc 2019; 16: 580-8. ArticlePubMedPMC
- 22. Wijsenbeek M, Cottin V. Spectrum of fibrotic lung diseases. N Engl J Med 2020; 383: 958-68. ArticlePubMed
- 23. Cottin V, Wollin L, Fischer A, Quaresma M, Stowasser S, Harari S. Fibrosing interstitial lung diseases: knowns and unknowns. Eur Respir Rev 2019; 28: 180100.ArticlePubMedPMC
- 24. Brown KK, Martinez FJ, Walsh SL, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J 2020; 55: 2000085.ArticlePubMedPMC
- 25. Nasser M, Larrieu S, Si-Mohamed S, et al. Progressive fibrosing interstitial lung disease: a clinical cohort (the PROGRESS study). Eur Respir J 2021; 57: 2002718.ArticlePubMedPMC
- 26. Flaherty KR, Brown KK, Wells AU, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res 2017; 4: e000212. ArticlePubMedPMC
- 27. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019; 381: 1718-27. ArticlePubMed
- 28. Maher TM, Corte TJ, Fischer A, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med 2020; 8: 147-57. ArticlePubMed
- 29. Behr J, Prasse A, Kreuter M, et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebocontrolled, phase 2b trial. Lancet Respir Med 2021; 9: 476-86. PubMed
- 30. Wells AU, Flaherty KR, Brown KK, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med 2020; 8: 453-60. PubMed
- 31. Matteson EL, Kelly C, Distler JH, et al. Nintedanib in patients with autoimmune disease-related progressive fibrosing interstitial lung diseases: subgroup analysis of the INBUILD trial. Arthritis Rheumatol 2022; 74: 1039-47. PubMedPMC
- 32. Sandoz E. Uber zwei falle von fotaler bronchektasie [About two cases of fetal bronchiectasis]. Beitr Pathol Anat 1907; 41: 496-517.
- 33. Marshall RP, Puddicombe A, Cookson WO, Laurent GJ. Adult familial cryptogenic fibrosing alveolitis in the United Kingdom. Thorax 2000; 55: 143-6. ArticlePubMedPMC
- 34. Hodgson U, Laitinen T, Tukiainen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax 2002; 57: 338-42. ArticlePubMedPMC
- 35. Barlo NP, van Moorsel CH, Ruven HJ, Zanen P, van den Bosch JM, Grutters JC. Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2009; 26: 155-61. PubMed
- 36. Garcia-Sancho C, Buendia-Roldan I, Fernandez-Plata MR, et al. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir Med 2011; 105: 1902-7. ArticlePubMed
- 37. Loyd JE. Pulmonary fibrosis in families. Am J Respir Cell Mol Biol 2003; 29: S47-50. PubMed
- 38. Fernandez BA, Fox G, Bhatia R, et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res 2012; 13: 64.ArticlePubMedPMCPDF
- 39. Ley B, Newton CA, Arnould I, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med 2017; 5: 639-47. ArticlePubMedPMC
- 40. Newton CA, Oldham JM, Ley B, et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J 2019; 53: 1801641.ArticlePubMedPMC
- 41. Cutting CC, Bowman WS, Dao N, et al. Family history of pulmonary fibrosis predicts worse survival in patients with interstitial lung disease. Chest 2021; 159: 1913-21. ArticlePubMedPMC
- 42. Steele MP, Speer MC, Loyd JE, et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med 2005; 172: 1146-52. ArticlePubMedPMC
- 43. Newton CA, Batra K, Torrealba J, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J 2016; 48: 1710-20. ArticlePubMedPMC
- 44. Mathai SK, Humphries S, Kropski JA, et al. MUC5B variant is associated with visually and quantitatively detected preclinical pulmonary fibrosis. Thorax 2019; 74: 1131-9. ArticlePubMed
- 45. Hunninghake GM, Quesada-Arias LD, Carmichael NE, et al. Interstitial lung disease in relatives of patients with pulmonary fibrosis. Am J Respir Crit Care Med 2020; 201: 1240-8. ArticlePubMedPMC
- 46. Salisbury ML, Hewlett JC, Ding G, et al. Development and progression of radiologic abnormalities in individuals at risk for familial interstitial lung disease. Am J Respir Crit Care Med 2020; 201: 1230-9. ArticlePubMedPMC
- 47. Mathai SK, Newton CA, Schwartz DA, Garcia CK. Pulmonary fibrosis in the era of stratified medicine. Thorax 2016; 71: 1154-60. ArticlePubMed
- 48. Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011; 364: 1503-12. PubMedPMC
- 49. van Moorsel CH, van der Vis JJ, Grutters JC. Genetic disorders of the surfactant system: focus on adult disease. Eur Respir Rev 2021; 30: 200085.ArticlePubMedPMC
- 50. Nogee LM, Dunbar AE, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med 2001; 344: 573-9. ArticlePubMed
- 51. Thomas AQ, Lane K, Phillips J 3rd, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med 2002; 165: 1322-8. ArticlePubMed
- 52. van Moorsel CH, Ten Klooster L, van Oosterhout MF, et al. SFTPA2 mutations in familial and sporadic idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2015; 192: 1249-52. ArticlePubMed
- 53. Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 2007; 356: 1317-26. ArticlePubMed
- 54. Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A 2007; 104: 7552-7. ArticlePubMedPMC
- 55. Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res 2012; 730: 52-8. ArticlePubMed
- 56. Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin North Am 2009; 23: 215-31. ArticlePubMedPMC
- 57. Diaz de Leon A, Cronkhite JT, Yilmaz C, et al. Subclinical lung disease, macrocytosis, and premature graying in kindreds with telomerase (TERT) mutations. Chest 2011; 140: 753-63. ArticlePubMedPMC
- 58. Borie R, Tabeze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J 2016; 48: 1721-31. ArticlePubMed
- 59. Juge PA, Borie R, Kannengiesser C, et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur Respir J 2017; 49: 1602314.ArticlePubMed
- 60. Petrovski S, Todd JL, Durheim MT, et al. An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am J Respir Crit Care Med 2017; 196: 82-93. ArticlePubMedPMC
- 61. Dressen A, Abbas AR, Cabanski C, et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: a candidate gene sequencing study. Lancet Respir Med 2018; 6: 603-14. ArticlePubMedPMC
- 62. Ley B, Torgerson DG, Oldham JM, et al. Rare protein-altering telomere-related gene variants in patients with chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med 2019; 200: 1154-63. ArticlePubMedPMC
- 63. Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med 2008; 178: 729-37. ArticlePubMedPMC
- 64. Leslie KO, Cool CD, Sporn TA, et al. Familial idiopathic interstitial pneumonia: histopathology and survival in 30 patients. Arch Pathol Lab Med 2012; 136: 1366-76. ArticlePubMedPDF
- 65. Viswanathan VS, Toro P, Corredor G, Mukhopadhyay S, Madabhushi A. The state of the art for artificial intelligence in lung digital pathology. J Pathol 2022; 257: 413-29. ArticlePubMedPMCPDF
- 66. Makela K, Mayranpaa MI, Sihvo HK, et al. Artificial intelligence identifies inflammation and confirms fibroblast foci as prognostic tissue biomarkers in idiopathic pulmonary fibrosis. Hum Pathol 2021; 107: 58-68. ArticlePubMed
- 67. Uegami W, Bychkov A, Ozasa M, et al. MIXTURE of human expertise and deep learning-developing an explainable model for predicting pathological diagnosis and survival in patients with interstitial lung disease. Mod Pathol 2022; 35: 1083-91. ArticlePubMedPMCPDF
- 68. Testa LC, Jule Y, Lundh L, et al. Automated digital quantification of pulmonary fibrosis in human histopathology specimens. Front Med (Lausanne) 2021; 8: 607720.ArticlePubMedPMC
- 69. Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol 1988; 41: 467-70. ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Identification of early genes in the pathophysiology of fibrotic interstitial lung disease in a new model of pulmonary fibrosis
Nathan Hennion, Corentin Bedart, Léonie Vandomber, Frédéric Gottrand, Sarah Humez, Cécile Chenivesse, Jean-Luc Desseyn, Valérie Gouyer
Cellular and Molecular Life Sciences.2025;[Epub] CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
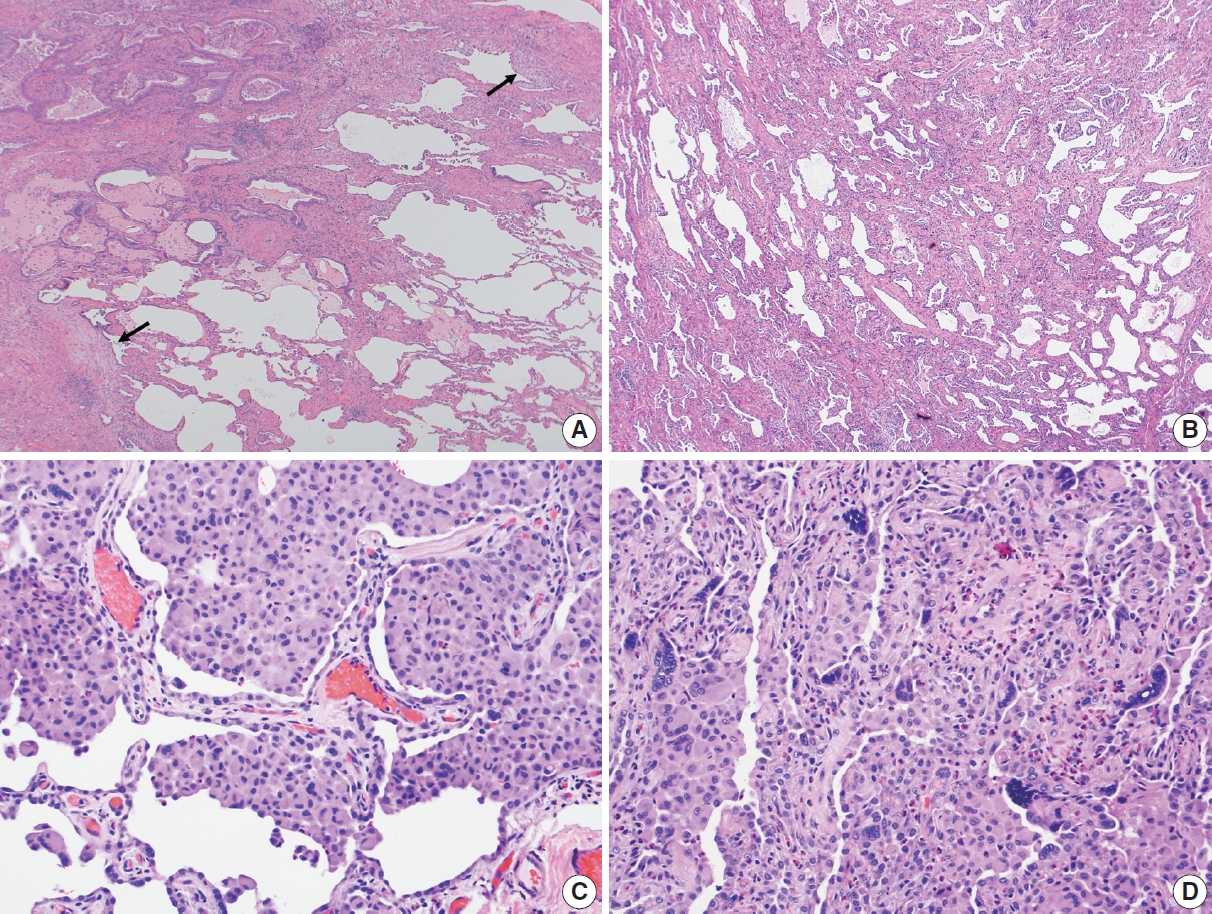
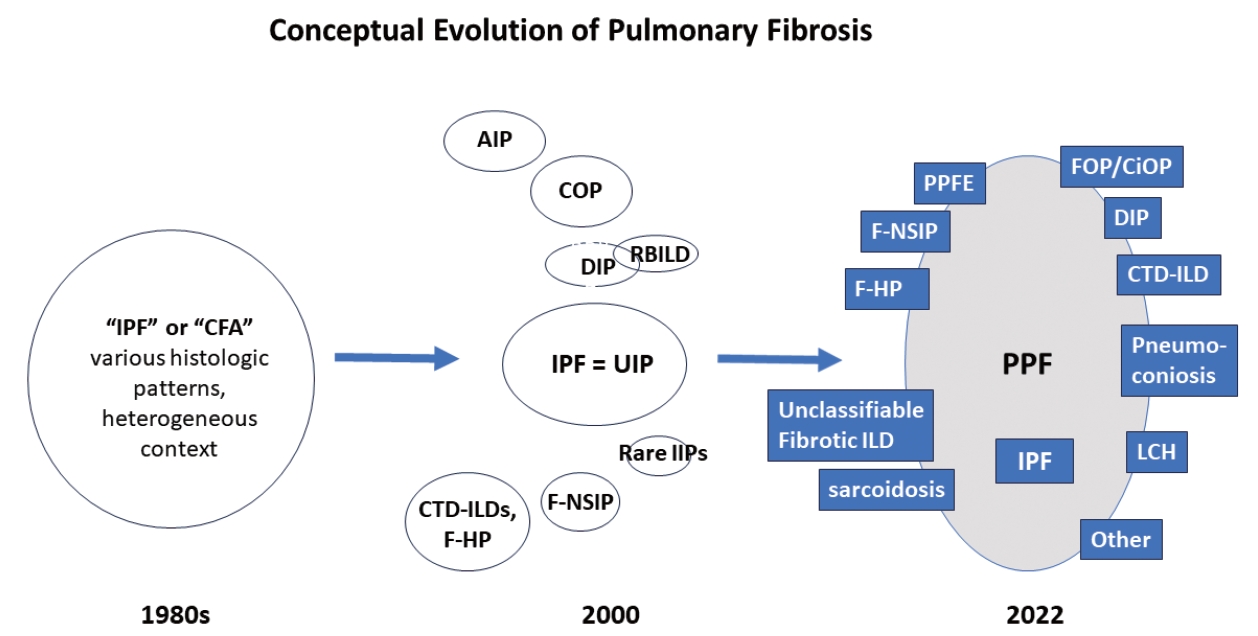



Fig. 1.
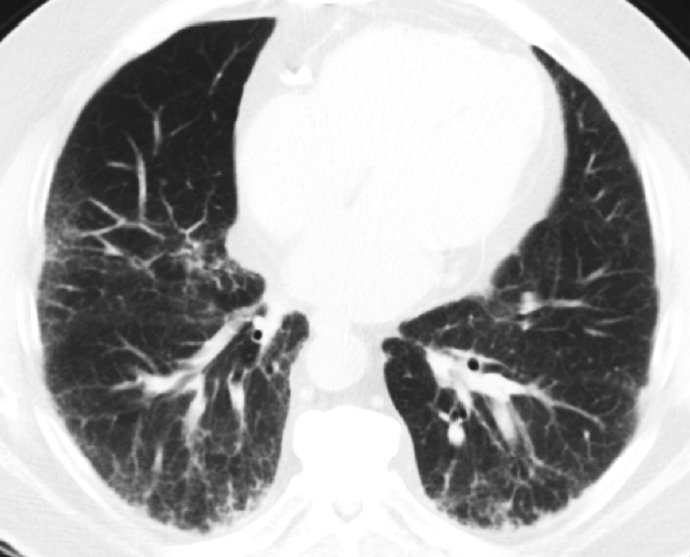
Fig. 2.
Fig. 3.
| Study | Purpose of study | Method | Country | No. of cases | Main finding |
|---|---|---|---|---|---|
| Makela et al. (2021) [66] | Quantitation of histologic parameters for survival analysis in IPF | Deep CNN | Finland | 71 IPF | Increased FF is associated with poor prognosis—interstitial mononuclear infiltrate & intra-alveolar macrophage with longer survival |
| Uegami et al. (2022) [67] | Prediction of UIP diagnosis | MIXTURE | Japan | 231 (UIP + non-UIP) cases; 715 WSI | A model approach to differentiate diseases by AI with collaboration with humans based on expert pathologists’ input |
| Testa et al. (2021) [68] | Pilot study to quantitate fibrosis in HPSPF & IPF | Automated quantitation of fibrosis with “dedicated software” | USA | 3 HPSPF & 9 IPF | Their automated image analysis offered accurate, reader-independent pulmonary fibrosis quantitation in HPSPF and IPF groups |
AI, artificial intelligence; CNN, convoluted neural network; IPF, idiopathic pulmonary fibrosis; FF, fibroblastic foci; MIXTURE, huMan-In-the loop eXpalainable artificial intelligence Through the Use of REcurrent training; UIP, usual interstitial pneumonia; WSI, whole slide imaging; HPSPF, Hermansky-Pudlak syndrome pulmonary fibrosis.

