E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 51(4); 2017 > Article
-
Review
Epstein-Barr Virus–Associated Lymphoproliferative Disorders: Review and Update on 2016 WHO Classification - Hyun-Jung Kim, Young Hyeh Ko1, Ji Eun Kim2, Seung-Sook Lee3, Hyekyung Lee4, Gyeongsin Park5, Jin Ho Paik6, Hee Jeong Cha7, Yoo-Duk Choi8, Jae Ho Han9, Jooryung Huh,10, Hematopathology Study Group of the Korean Society of Pathologists
-
Journal of Pathology and Translational Medicine 2017;51(4):352-358.
DOI: https://doi.org/10.4132/jptm.2017.03.15
Published online: June 5, 2017
Department of Pathology, Inje University, Sanggye Paik Hospital, Seoul, Korea
1Sungkyunkwan University, School of Medicine, Samsung Medical Center, Seoul, Korea
2SMG-SNU Boramae Medical Center, Seoul National University, Seoul, Korea
3Korea Cancer Center Hospital, Seoul, Korea
4Eulji University Hospital, Eulji University School of Medicine, Daejeon, Korea
5Gangnam St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea
6Seoul National University Bundang Hospital, Seongnam, Korea
7Ulsan University Hospital, Ulsan University School of Medicine, Ulsan, Korea
8Chonnam National University Hospital, Chonnam National University, Gwangju, Korea
9Ajou University Hospital, Suwon,Korea
10Asan Medical Center, Ulsan University College of Medicine, Seoul, Korea
- Corresponding Author Jooryung Huh, MD Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea Tel: +82-2-3010-4553 Fax: +82-2-472-7898 E-mail: 'Jrhuh@amc.seoul.kr'
© 2017 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Epstein-Barr virus (human herpesvirus-4) is very common virus that can be detected in more than 95% of the human population. Most people are asymptomatic and live their entire lives in a chronically infected state (IgG positive). However, in some populations, the Epstein-Barr virus (EBV) has been involved in the occurrence of a wide range of B-cell lymphoproliferative disorders (LPDs), including Burkitt lymphoma, classic Hodgkin’s lymphoma, and immune–deficiency associated LPDs (post-transplant and human immunodeficiency virus–associated LPDs). T-cell LPDs have been reported to be associated with EBV with a subset of peripheral T-cell lymphomas, angioimmunoblastic T-cell lymphomas, extranodal nasal natural killer/T-cell lymphomas, and other rare histotypes. This article reviews the current evidence covering EBV-associated LPDs based on the 2016 classification of the World Health Organization. These LPD entities often pose diagnostic challenges, both clinically and pathologically, so it is important to understand their unique pathophysiology for correct diagnoses and optimal management.
- The spectrum of EBV-associated B-cell LPDs is broad, ranging from reactive lymphoproliferative lymphadenitis to lymphomas. All the related disease entities are shown in Table 1. Well-defined lymphoma entities such as Burkitt lymphoma, classical Hodgkin lymphoma, and plasmablastic lymphoma are not included [4].
- Infectious mononucleosis
- Primary EBV infection occurs most often in childhood and is generally asymptomatic. In adolescence, it is associated with a self-limiting infectious mononucleosis syndrome, manifested by fever, pharyngitis, malaise, and atypical lymphocytosis. Following primary infection, most individuals remain a life-long carrier of the virus without serious sequelae [5]. However, a small population with the latent infection will develop various LPDs.
- EBV is transmitted from the host by saliva and infected EBV replicates within oropharyngeal epithelium and is then exposed to circulating B-lymphocytes. Peripheral EBV-infected memory B-cells can return to the Waldeyer’s ring, undergoing reactivation to produce an infectious virus that will be shed in the saliva. EBV-specific cytotoxic T-cells (CTL) destroy most infected cells.
- The histologic features of infectious mononucleosis vary during the course of the disease. Early in the disorder, follicular hyperplasia occurs with monocytoid B-cell aggregates and epithelioid histiocytes. Later, the expansion of the paracortex predominates. The immunoblasts resemble classical Reed-Sternberg (RS) cells. The immunoblasts of predominantly B-cell types and partly T-cell types often express CD30. In-situ hybridization (ISH) of EBV-encoded small RNAs (EBERs) exhibit numerous positive immunoblasts in the paracortex but not in the germinal centers [6].
- Chronic active EBV of B-cell types
- As first defined by Lekstrom-Himes et al. [7], chronic active EBV (CAEBV) of B-cell types refers to a chronic or persistent EBV infection characterized by a severe illness lasting more than 6 months, persistent elevated EBV titers, and evidence of EBV-related organ damage. Currently, CAEBV is defined as (1) a severe progressive illness with a duration of more than 6 months, (2) lymphocytic infiltration of tissue (e.g., lymph nodes, lungs, liver, central nervous tissue, bone marrow, eye, and skin), (3) elevated EBV DNA and RNA in affected tissue, and (4) absence of any other immunosuppressive conditions [8].
- Histologically, the lymph nodes exhibits features resembling polymorphic PTLD, with paracortical expansion, plasmacytoid lymphoblastic proliferation, presence of plasma cells, and presence of occasional RS-like cells. EBV-ISH positive B-cells are noted in the paracortex. Among CAEBV patients, 63% had clonal immunoglobulin rearrangement [9].
- EBV-positive diffuse large B-cell lymphoma
- EBV-positive diffuse large B-cell lymphoma (DLBCL), not otherwise specified was originally described as “senile EBV-associated B-cell LPD,” or “EBV-positive DLBCL of the elderly” (older than 50 years) (WHO 4th edition) [10]. However, subsequent studies have shown that EBV-positive DLBCL is not limited to this older age group. In the elderly group, it is thought to be related to immunosenescence, which modifies T-cell homeostasis through a lack of thymic output of naïve T-cells and an accumulation of viral specific CD8+ T cells.
- Histologically, four types have been described: monomorphic (DLBCL-like, monotonous sheets of large cells), polymorphic in the inflammatory background, T-cell/histiocyte-rich large cell lymphoma, and plasmacytoid differentiation. Immunophenotypically, the tumor cells express pan B-cell markers (CD20, PAX5, CD79a, OCT-2, and BOB-1), and are mostly CD30+ , but lack CD15 expression.
- This disease entity frequently involves extranodal sites including the skin, lung, tonsils, and stomach. Unfavorable prognostic factors include older age (>70 years), high international prognostic index, and activated B-cell phenotype [11].
- EBV mucocutaneous ulcer
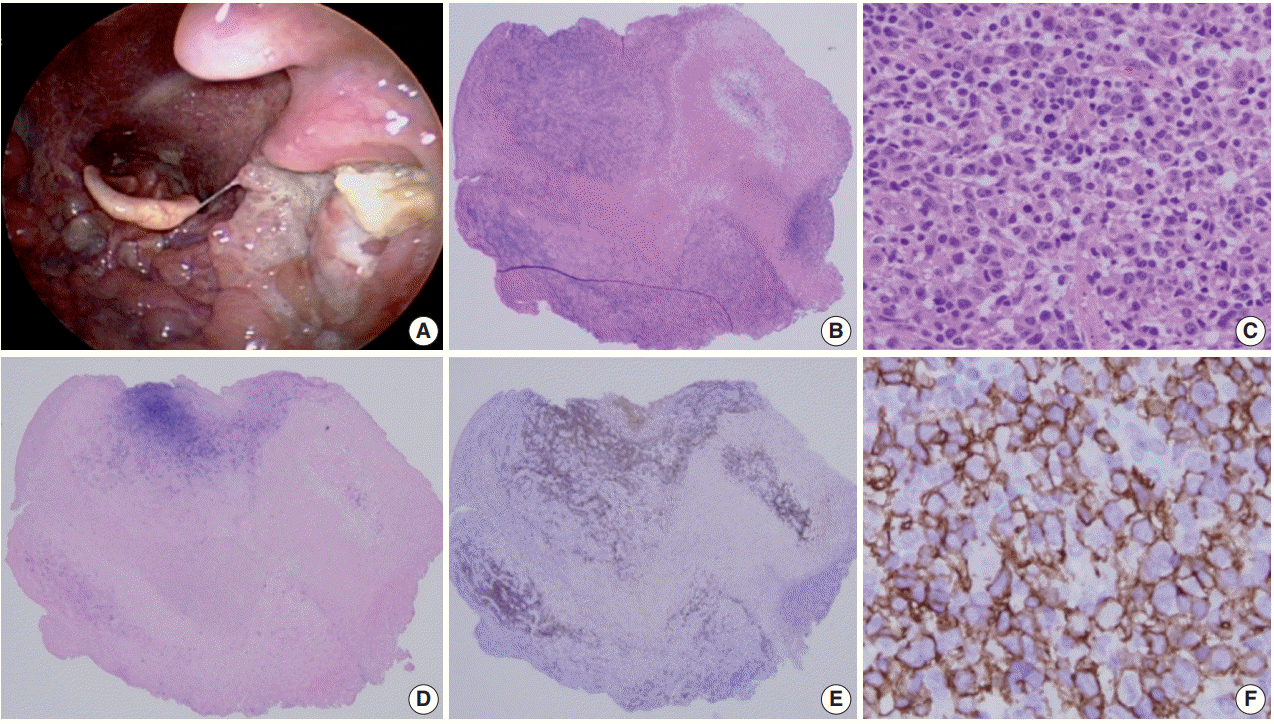
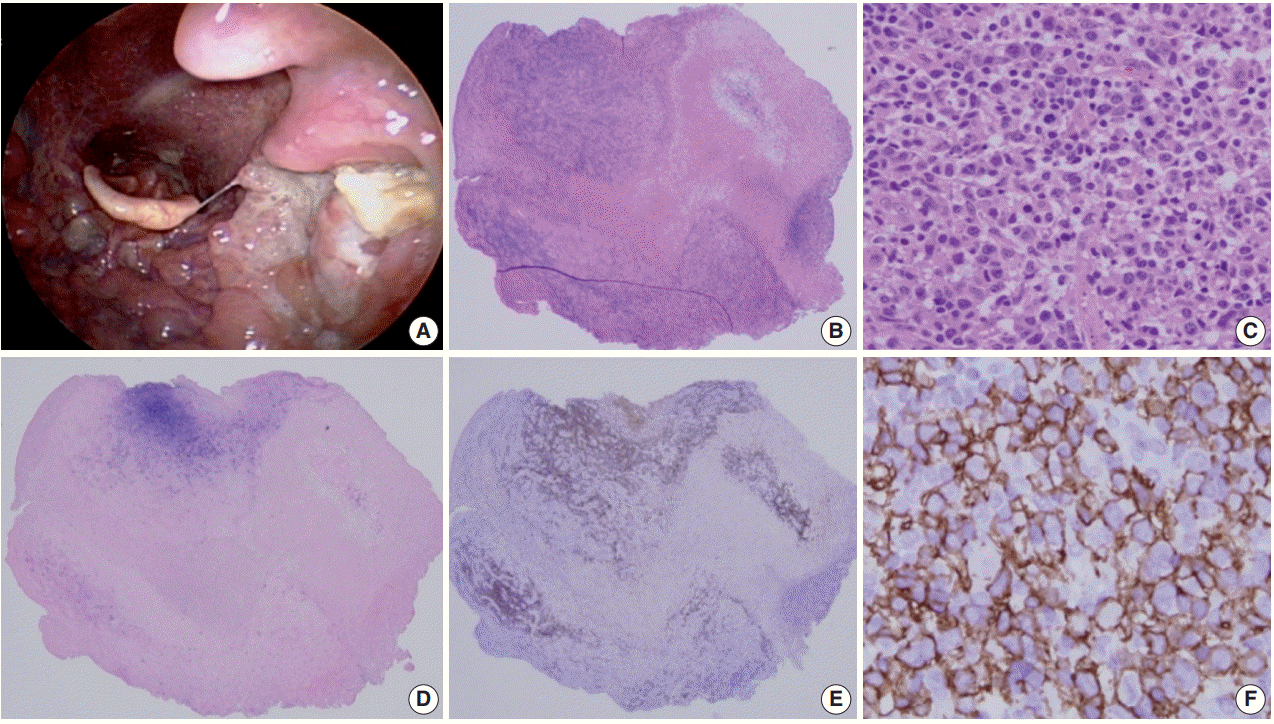
- This self-limited EBV positive B-cell proliferation is characterized by the presence of mucocutaneous ulcers with an indolent clinical course. This phenomenon is likely due to a more localized form of decreased immune surveillance, which is supported by a very low EBV viral load. The frequently involved sites includes the skin, oropharyngeal mucosa, and gastrointestinal tract [12].
- Histologically a sharply demarcated ulcer is lined by an inflammatory infiltrate with clusters of large atypical cells, often with RS cell-like features. Phenotypically the large atypical cells are variably positive for CD20 and CD30, and uniformly positive for EBV-ISH and CD15+ in half of the cases (Fig. 1).
- DLBCL associated with chronic inflammation
- DLBCL associated with chronic inflammation develops in the setting of long-standing chronic inflammation with EBV association. It usually involves body cavities or enclosed spaces (like cysts). Pyothorax-associated lymphoma (PAL) represents the prototype of this entity.
- Cases with PAL have long history of chronic pyothorax and may present with chest pain, fever, cough, dyspnea, and tumor mass. The prognosis of PAL is poor.
- The morphology discloses a diffuse proliferation of large atypical lymphocytes with plasmacytoid cytomorphology. Immunohistochemistry reveals neoplastic cells that represent pan-B cell markers, usually positive for IRF4/MUM1, and CD13. An aberrant expression of T-cell phenotypes is also seen.
- The unique genomic instability has been reported as follows: A20 deletion, interferon-inducible 27 (IFI27), TP53 mutation, and MYC amplification [13].
- Lymphomatoid granulomatosis
- Katzenstein et al. [14] initially described a rare angiocentric and angiodestructive EBV-associated LPD, which was distinct from Wegener’s granulomatosis.
- Nearly all patients present with symptoms related to pulmonary involvement, followed by involved sites of the central nervous system, skin, liver, and kidney. Radiologically, bilateral variable sized lung nodules are noted in lymphomatoid granulomatosis (LYG).
- The histologic features of LYG are observed in lung nodules. All the lesions are angioinvasive and angiocentric with fibrinoid necrosis of the vascular wall. The infiltrate is polymorphous with an admixture of small lymphocytes, histiocytes, and large lymphoid cells. The grading of LYG is based on the proportion of EBV positive cells. In general, grade 1 and grade 2 lesions are approached using strategies that are designed to improve the host’s immune system, whereas grade 3 lesions require chemotherapy and do not respond to immunomodulatory therapies. Phenotypically, large atypical EBV-positive B cells express CD20, PAX5, CD79a, CD30+ , and CD15– [15].
- EBV-ASSOCIATED T-CELL AND NATURAL KILLER CELL LPDS
- EBV is a ubiquitous herpes virus with tropism for B cells, but the infection of T cells and natural killer (NK) cells may lead to several EBV-related LPDs. EBV-positive T/NK LPD encompasses disease entities with a broad clinicopathologic spectrum (Table 2) [16].
- EBV-associated hemophagocytic lymphohistiocytosis
- Hemophagocytic lymphohistiocytosis (HLH) is a clinicopathologic syndrome encompassing a markedly dysregulated immune response and hypercytokinemia. HLH is characterized clinically by fever, splenomegaly, and cytopenias, and histologically by hemophagocytosis. The supportive laboratory findings for HLH are as follows: extremely high serum level of ferritin, lactate dehydrogenase, soluble CD25, and elevated viral capsid. EBV associated HLH accounts for 40% of HLH.
- Histologically, EBV-positive T-cells and hemophagocytosing histiocytes are scattered in the sinusoids of the bone marrow and liver. The T-cells express CD8 and granzyme B. Even though it is uncommon, NK cells can be infiltrated in HLH [17].
- HLH can be effectively controlled in most patients (more than 90%), but the other 10% often die of fulminant disease.
- CAEBV infection of T-cell or NK-cell types, systemic
- CAEBV infection was initially defined as follows: (1) markedly abnormal EBV antibody titer; (2) histologic evidence of organ involved- interstitial pneumonia, hypoplasia of the bone marrow, uveitis, lymphadenitis, persistent hepatitis, or splenomegaly; and (3) increased EBV RNA in affected tissue [18].
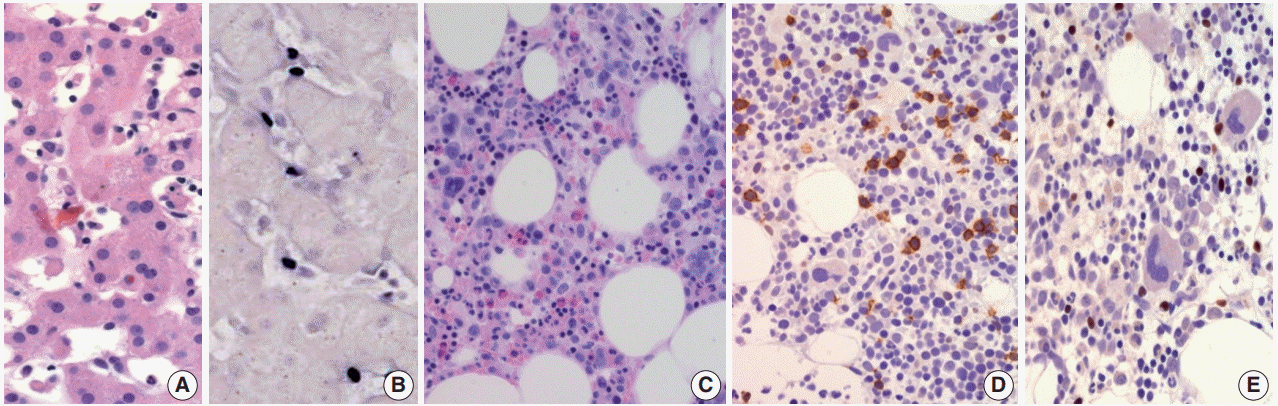
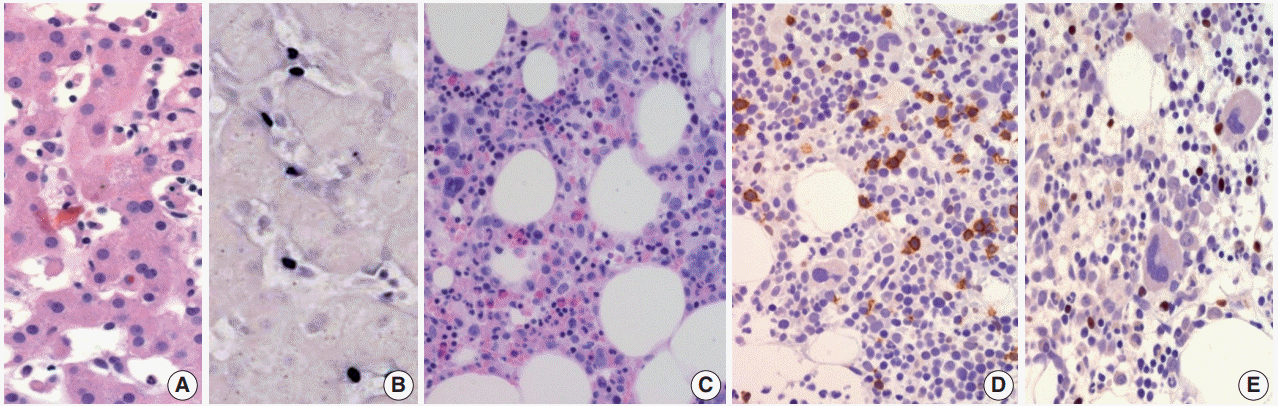
- Clinically, CAEBV-T/NK is a disease of children but is also detected in young adults and even in middle-aged older adults with a mean age of 11.3 years [19]. The symptoms are usually prolonged fever, hepatomegaly, splenomegaly, thrombocytopenia, anemia, and lymphadenopathy [19]. Life-threatening complications include hemophagocytic syndrome, interstitial pneumonia, malignant lymphoma, coronary aneurysms, and central nervous system involvement. All patients have elevated levels of EBV DNA in their blood, which is well-correlated with clinical severity [19].
- Morphologically, in patients with CAEBV-T/NK the lymph nodes exhibit paracortical hyperplasia with polymorphic and polyclonal lymphoid proliferation and large numbers of EBER positive cells. The liver exhibits portal or sinusoidal infiltration by small lymphocytes with no definite atypia (Fig. 2).
- Ohshima et al. [18] proposed a three-tier classification as follows: category A1 is polymorphic LPD with polyclonal proliferation of EBV-infected T cells or NK cells; category A2 is polymorphic LPD with monoclonal T/NK cells; and category A3 is monomorphic LPD of monoclonal T cells.
- Severe mosquito bite allergy
- A severe mosquito bite allergy is a cutaneous manifestation of chronic EBV infection characterized by intense local skin symptoms, such as erythema, bullae, ulcers, and scarring. The systemic symptoms such as fever, lymphadenopathy, and liver dysfunction are developed after mosquito bites, vaccination, or injection.
- The epidermis at the mosquito bite site exhibits necrosis and ulceration. The dermis reveals edema and infiltration of polymorphonuclear leukocytes, nuclear debris, and extravasated erythrocytes with fibrinoid necrosis of small vessels. The infiltrating small lymphocytes extend from the dermis to the subcutis in an angiocentric pattern. EBV-positive cells represent 3%–10% of infiltrating lymphocytes [20].
- Hydroa vacciniforme-like LPD
- Hydroa vacciniforme (HV)-like LPD is one of the cutaneous forms of CAEBV. It is initially described as an EBV positive polyclonal or monoclonal T/NK LPD, characterized by blistering photodermatoses in childhood and healed with vacciniform scarring [21]. It is clinically divided into two types. The classic type is a self-limited disease with vesicles on sun-exposed areas in adolescence or young adulthood. Severe HV-type tends to exhibit more extensive skin lesions and systemic manifestations of fever, hepatomegaly, serologic abnormalities, and peripheral NK lymphocytosis. The severe type of HV often progresses to EBV-associated NK/T-cell malignancy [22,23].
- Morphologic findings of HV are epidermal reticular degeneration to spongiotic vesiculation with perivascular and periappendiceal lymphocytic infiltration with no definite cytologic atypia. Severe HV and HV-like T-cell lymphomas mimic those of classic HV, but the dermal infiltrates are more extensive and deeper, composed of variably atypical lymphocytes.
- The immunophenotype of the classic HV is CD4+ or CD8+ T-cells, but the severe form/lymphoma exhibits predominantly CD8+ CTLs. The majority are αβ T cells. A few cases involve αβ T cells and rarely NK cells.
- Systemic EBV-positive T-cell lymphoma
- Systemic EBV-positive T-cell lymphoma of childhood and young adulthood is a fulminant illness of EBV-infected T cells with clonal proliferation and cytotoxic phenotype. The clinical manifestation of this lesion is a rapid clinical progression with multiple organ failure, sepsis, and death. A hemophagocytic syndrome is nearly always associated. Systemic EBV-positive T-cell lymphoma arising in patients with a history of CAEBV-T/NK develops in a median time of 35 months [24].
- Hyperplasia of histiocytes and marked hemophagocytic syndrome are also noted with increased small T-cells in the bone marrow, spleen, and liver. The paracortical zone of the lymph node is expanded with the depletion of B-cell areas. The degree of cytologic atypia in EBV-positive lymphocytes is variable.
- Extranodal NK/T cell lymphoma, nasal type
- Extranodal NK/T cell lymphoma is a prototype of EBV-associated T-cell LPD, which is characterized by frequent necrosis, angiocentric growth, cytotoxic phenotype and a strong association with EBV. Since the nasal cavity is the most commonly involved site, the nasopharynx and upper aerodigestive areas including the nasal cavity disclose progressively destructive and ulcerative lesions or obstructive symptoms due to mass effects [25].
- Extranasal NK/T cell lymphoma
- Extranasal NK/T cell lymphomas frequently involve the skin, gastrointestinal tract, testis, and soft tissue. Most patients present at a higher stage with multiple areas of involvement [26].
- The morphology of the involved sites is frequently ulcerated and necrotic. The cytologic composition varies ranging from small, medium, and large. Angiocentric growth accompanies diffuse necrosis, and vascular damage.
- Aggressive NK cell leukemia
- Aggressive NK-cell leukemia is a neoplasm of NK cells, which primarily involves peripheral blood and bone marrow. In contrast to conventional leukemia, the tumor cells may not be abundant in the peripheral blood and bone marrow. The average age of patients exhibiting this disorder is 39 years. The typical presentations include fever, hepatosplenomegaly, lymphadenopathy, and is complicated by hemophagocytic syndrome.
- In histologic sections, there are diffuse, destructive and permeative infiltrates of monomorphic cells with a round to moderate rim of pale or amphophilic cytoplasm. Interspersed apoptotic bodies and zonal cell death are common. Angioinvasive and angiodestructive growth is also frequently noted. The clinical course is fatal.
- EBV-positive nodal NK/T cell lymphoma
- EBV-positive nodal T/NK cell lymphoma may involve a limited number of extranodal organs except for the nasal cavity, but the main bulk of the tumor is located in the lymph nodes. This entity is very rare with fewer than 100 cases being reported in the literature [27].
- The lymph nodes exhibit a diffuse infiltration of pleomorphic, variable sized cells. The cytomorphology of the tumor cells are more commonly centroblastoid, often anaplastic or plasmacytoid with some RS-like cells being noted. Some cases show extensive necrosis, many apoptotic bodies, and angiocentric growth patterns. The immunophenotype is as follows: CD3+, CD8+, TIA+, and granzyme B+ [28].
- The disease course is very aggressive, with a median survival of only 4 months.
EPSTEIN-BARR VIRUS–ASSOCIATED B-CELL LYMPHOPROLIFERATIVE DISORDERS
- Many new entities and concepts for EBV-positive LPDs have been added to the 2016 WHO classification, based on growing knowledge in the field of genetics and molecular virology. With this increased understanding of LPD the clinical and pathologic entities, we can perform EBV-ISH indicative of (1) clinically bordered between infection and neoplastic conditions; (2) past history of recurrent inappropriate immune response, especially in children/young adults or old age; and (3) a pathologically polymorphous inflammatory background (but not Hodgkin lymphoma). It helps that the final destination is the achievement of appropriate diagnosis and management of LPDs. Some entities are provisional and must wait for confirmation of additional data.
CONCLUSION
-
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Notes
- 1. Cohen JI. Epstein-Barr virus infection. N Engl J Med 2000; 343: 481–92. ArticlePubMed
- 2. Pittaluga S, Said J. Virally associated B cell lymphoproliferative disease. In : Jaffe E, Arbor DA, Campo E, Harris NL, Quintanilla-Fend L, eds. Hematopathology. 2nd ed. Philadelphia: Elsevier Heealth Science, 2016; 547–607.
- 3. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127: 2375–90. ArticlePubMedPMC
- 4. Chaganti S, Heath EM, Bergler W, et al. Epstein-Barr virus colonization of tonsillar and peripheral blood B-cell subsets in primary infection and persistence. Blood 2009; 113: 6372–81. ArticlePubMed
- 5. Vouloumanou EK, Rafailidis PI, Falagas ME. Current diagnosis and management of infectious mononucleosis. Curr Opin Hematol 2012; 19: 14–20. ArticlePubMed
- 6. Fletcher C. Diagnostic histopathology of tumors. 3rd ed. Philadelphia: Churchill Livingstone/Elsevier, 2007; 1260–1.
- 7. Lekstrom-Himes JA, Dale JK, Kingma DW, Diaz PS, Jaffe ES, Straus SE. Periodic illness associated with Epstein-Barr virus infection. Clin Infect Dis 1996; 22: 22–7. ArticlePubMed
- 8. Cho EY, Kim KH, Kim WS, Yoo KH, Koo HH, Ko YH. The spectrum of Epstein-Barr virus-associated lymphoproliferative disease in Korea: incidence of disease entities by age groups. J Korean Med Sci 2008; 23: 185–92. ArticlePubMedPMC
- 9. Cohen JI, Jaffe ES, Dale JK, et al. Characterization and treatment of chronic active Epstein-Barr virus disease: a 28-year experience in the United States. Blood 2011; 117: 5835–49. ArticlePubMedPMC
- 10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumors of hematopoietic and lymphoid tissues. 4th ed. Lyon: IARC Press, 2008.
- 11. Nicolae A, Pittaluga S, Abdullah S, et al. EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood 2015; 126: 863–72. ArticlePubMedPMC
- 12. Dojcinov SD, Venkataraman G, Raffeld M, Pittaluga S, Jaffe ES. EBV positive mucocutaneous ulcer--a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol 2010; 34: 405–17. ArticlePubMedPMC
- 13. Hongyo T, Kurooka M, Taniguchi E, et al. Frequent p53 mutations at dipyrimidine sites in patients with pyothorax-associated lymphoma. Cancer Res 1998; 58: 1105–7. PubMed
- 14. Katzenstein AL, Carrington CB, Liebow AA. Lymphomatoid granulomatosis: a clinicopathologic study of 152 cases. Cancer 1979; 43: 360–73. ArticlePubMed
- 15. Song JY, Pittaluga S, Dunleavy K, et al. Lymphomatoid granulomatosis: a single institute experience: pathologic findings and clinical correlations. Am J Surg Pathol 2015; 39: 141–56. ArticlePubMedPMC
- 16. Kimura H, Ito Y, Kawabe S, et al. EBV-associated T/NK-cell lymphoproliferative diseases in nonimmunocompromised hosts: prospective analysis of 108 cases. Blood 2012; 119: 673–86. ArticlePubMed
- 17. Toga A, Wada T, Sakakibara Y, et al. Clinical significance of cloned expansion and CD5 down-regulation in Epstein-Barr Virus (EBV)-infected CD8+ T lymphocytes in EBV-associated hemophagocytic lymphohistiocytosis. J Infect Dis 2010; 201: 1923–32. ArticlePubMed
- 18. Ohshima K, Kimura H, Yoshino T, et al. Proposed categorization of pathological states of EBV-associated T/natural killer-cell lymphoproliferative disorder (LPD) in children and young adults: overlap with chronic active EBV infection and infantile fulminant EBV T-LPD. Pathol Int 2008; 58: 209–17. ArticlePubMed
- 19. Ko YH, Kim HJ, Oh YH, et al. EBV-associated T and NK cell lymphoproliferative disorders: consensus report of the 4th Asian Hematopathology Workshop. J Hematopathol 2012; 5: 319–24. Article
- 20. Cohen JI, Kimura H, Nakamura S, Ko YH, Jaffe ES. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol 2009; 20: 1472–82. ArticlePubMedPMC
- 21. Park S, Ko YH. Epstein-Barr virus-associated T/natural killer-cell lymphoproliferative disorders. J Dermatol 2014; 41: 29–39. ArticlePubMed
- 22. Miyake T, Yamamoto T, Hirai Y, et al. Survival rates and prognostic factors of Epstein-Barr virus-associated hydroa vacciniforme and hypersensitivity to mosquito bites. BrJ Dermatol 2015; 172: 56–63. Article
- 23. Cho KH, Kim CW, Heo DS, et al. Epstein-Barr virus-associated peripheral T-cell lymphoma in adults with hydroa vacciniforme-like lesions. Clin Exp Dermatol 2001; 26: 242–7. ArticlePubMed
- 24. Kumar S, et al. FulminantEBV(+)T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood 2000; 96: 443–51. ArticlePubMedPDF
- 25. Hong M, Lee T, Kang SY, Kim SJ, Kim W, Ko YH. Nasal-type NK/T-cell lymphomas are more frequently T rather than NK lineage based on T-cell receptor gene, RNA, and protein studies: lineage does not predict clinical behavior. Mod Pathol 2016; 29: 430–43. ArticlePubMed
- 26. Chan JK, Sin VC, Wong KF, et al. Nonnasal lymphoma expressing the natural killer cell marker CD56: a clinicopathologic study of 49 cases of an uncommon aggressive neoplasm. Blood 1997; 89: 4501–13. ArticlePubMedPDF
- 27. Kato S, Asano N, Miyata-Takata T, et al. T-cell receptor (TCR) phenotype of nodal Epstein-Barr virus (EBV)-positive cytotoxic T-cell lymphoma (CTL): a clinicopathologic study of 39 cases. Am J Surg Pathol 2015; 39: 462–71. ArticlePubMed
- 28. Kim JH, Sung JY, Han JH, KoYH; Hematopathology Study Group of the Korean Society of Pathologists. Epstein-Barr virus-positive nodal T/NK-cell lymphoma: an analysis of 15 cases with distinct clinicopathological features. Hum Pathol 2015; 46: 981–90. ArticlePubMed
References
Figure & Data
References
Citations
- Epstein–Barr virus‐positive monoclonal lymphoplasmacytic proliferation associated with neurosyphilis in an immunocompetent patient: A case report
Takashi Hibiya, Kiyotaka Nagahama, Yoshie Matsumoto, Kuniaki Saito, Nobuyoshi Sasaki, Keiichi Kobayashi, Akiyasu Otsu, Teppei Shimasaki, Kengo Takeuchi, Yoshiaki Shiokawa, Motoo Nagane, Junji Shibahara
Neuropathology.2024; 44(2): 104. CrossRef - Epstein-Barr virus-positive iris diffuse large B-cell lymphoma detected by metagenomic next-generation sequencing
Xiao-na Wang, Jing Hong, Yong-gen Xu, Pei Zhang, Ying-yu Li, Hong-liang Dou, Hai-ping Li
BMC Ophthalmology.2024;[Epub] CrossRef - Pharmacological Modulation of the Crosstalk between Aberrant Janus Kinase Signaling and Epigenetic Modifiers of the Histone Deacetylase Family to Treat Cancer
Al-Hassan M. Mustafa, Oliver H. Krämer, Lynette Daws
Pharmacological Reviews.2023; 75(1): 35. CrossRef - Autophagy-associated immune dysregulation and hyperplasia in a patient with compound heterozygous mutations in ATG9A
Guowu Hu, Pia J Hauk, Nannan Zhang, Waleed Elsegeiny, Carlos M. Guardia, Amy Kullas, Kevin Crosby, Robin R. Deterding, Michaela Schedel, Paul Reynolds, Jordan K Abbott, Vijaya Knight, Stefania Pittaluga, Mark Raffeld, Sergio D. Rosenzweig, Juan S. Bonifac
Autophagy.2023; 19(2): 678. CrossRef - When to suspect inborn errors of immunity in Epstein–Barr virus–related lymphoproliferative disorders
Keith A. Sacco, Luigi D. Notarangelo, Ottavia M. Delmonte
Clinical Microbiology and Infection.2023; 29(4): 457. CrossRef - Primary head and neck cancer cell cultures are susceptible to proliferation of Epstein-Barr virus infected lymphocytes
Senyao Shao, Lars Uwe Scholtz, Sarah Gendreizig, Laura Martínez-Ruiz, Javier Florido, Germaine Escames, Matthias Schürmann, Carsten Hain, Leonie Hose, Almut Mentz, Pascal Schmidt, Menghang Wang, Peter Goon, Michael Wehmeier, Frank Brasch, Jörn Kalinowski,
BMC Cancer.2023;[Epub] CrossRef - Clinical and genetic characterization of Epstein-Barr virus–associated T/NK-cell lymphoproliferative diseases
Hui Luo, Dan Liu, Wenbing Liu, Jin Jin, Xiaoman Bi, Peiling Zhang, Jia Gu, Miao Zheng, Min Xiao, Xin Liu, Jianfeng Zhou, Qian-Fei Wang
Journal of Allergy and Clinical Immunology.2023; 151(4): 1096. CrossRef - Outcomes of programmed death protein-1 inhibitors treatment of chronic active Epstein Barr virus infection: A single center retrospective analysis
Yaxian Ma, Peiling Zhang, Yuhan Bao, Hui Luo, Jiachen Wang, Liang Huang, Miao Zheng
Frontiers in Immunology.2023;[Epub] CrossRef - Pathogenesis, treatment and prevention of diseases caused by Epstein–Barr virus
A. G. Rumyantsev
Pediatric Hematology/Oncology and Immunopathology.2023; 22(2): 166. CrossRef - Epstein–Barr virus-associated B-cell lymphoproliferative disorder meeting the definition of CAEBV B cell disease: a case report
Yaxian Ma, Yuhan Bao, Miao Zheng
BMC Infectious Diseases.2023;[Epub] CrossRef - Unpacking the CNS Manifestations of Epstein-Barr Virus: An Imaging Perspective
N. Soni, M. Ora, R. Singh, P. Mehta, A. Agarwal, G. Bathla
American Journal of Neuroradiology.2023; 44(9): 1002. CrossRef - Oncoviruses: Induction of cancer development and metastasis by increasing anoikis resistance
Zahra Sobhi Amjad, Ali Shojaeian, Javid Sadri Nahand, Mobina Bayat, Mohammad Taghizadieh, Mosayeb Rostamian, Farhad Babaei, Mohsen Moghoofei
Heliyon.2023; 9(12): e22598. CrossRef - Frequency and association of Epstein-Barr Virus genotype in rheumatoid arthritis patients of Khyber Pakhtunkhwa, Pakistan
Ayesha Munir, Suleman Khan, Sanaullah Khan, Sobia Attaullah, Mehwish Munir, Aisha Saleem, Ijaz Ali, Hideo Kato
PLOS ONE.2023; 18(12): e0295124. CrossRef - Successful treatment by using a modified SMILE regimen and autologous hematopoietic stem cell transplantation in a pediatric primary EBV-positive nodular NK/T cell lymphoma patient
Jian Li, Juxin Ye, Yongren Wang, Jun Wang, Yongjun Fang
Annals of Hematology.2022; 101(2): 433. CrossRef - Genetic errors of immunity distinguish pediatric nonmalignant lymphoproliferative disorders
Lisa R. Forbes, Olive S. Eckstein, Nitya Gulati, Erin C. Peckham-Gregory, Nmazuo W. Ozuah, Joseph Lubega, Nader K. El-Mallawany, Jennifer E. Agrusa, M. Cecilia Poli, Tiphanie P. Vogel, Natalia S. Chaimowitz, Nicholas L. Rider, Emily M. Mace, Jordan S. Ora
Journal of Allergy and Clinical Immunology.2022; 149(2): 758. CrossRef - EBV-positive B-cell ulcerative proliferation in the oral cavity associated with EBV-negative follicular lymphoma in a patient with common variable immunodeficiency: A case report and review of the literature
Waleed A. Alamoudi, Antoine Azar, Stefan K. Barta, Faizan Alawi, Takako I. Tanaka, Eric T. Stoopler, Thomas P. Sollecito
Oral Surgery, Oral Medicine, Oral Pathology and Oral Radiology.2022; 133(1): e10. CrossRef - Necrotizing Follicular Lymphoma of the Inguinal Region with Sternbergoid Cells: Clinical–Pathological Features of a Challenging Entity
Federico Scarmozzino, Marco Pizzi, Marta Sbaraglia, Luisa Santoro, Luca Frison, Silvia Nalio, Laura Bonaldi, Livio Trentin, Angelo Paolo Dei Tos
Applied Sciences.2022; 12(3): 1290. CrossRef - High percentages of peripheral blood T-cell activation in childhood Hodgkin's lymphoma are associated with inferior outcome
Fengqing Cai, Hui Gao, Zhongsheng Yu, Kun Zhu, Weizhong Gu, Xiaoping Guo, Xiaojun Xu, Hongqiang Shen, Qiang Shu
Frontiers in Medicine.2022;[Epub] CrossRef - Case Report of a Novel NFkB Mutation in a Lymphoproliferative Disorder Patient
Khashayar Danandeh, Parnian Jabbari, Elham Rayzan, Samaneh Zoghi, Sepideh Shahkarami, Raul Jimenez Heredia, Ana Krolo, Bibi Shahin Shamsian, Kaan Boztug, Nima Rezaei
Endocrine, Metabolic & Immune Disorders - Drug Targets.2022; 22(10): 1040. CrossRef - EBV-associated diseases: Current therapeutics and emerging technologies
Srishti Chakravorty, Behdad Afzali, Majid Kazemian
Frontiers in Immunology.2022;[Epub] CrossRef - Clinical features and treatment strategies for post-transplant and iatrogenic immunodeficiency-associated lymphoproliferative disorders
Akihiro Ohmoto, Shigeo Fuji
Blood Reviews.2021; 49: 100807. CrossRef - Comparative Study on Epstein-Barr Virus-Positive Mucocutaneous Ulcer and Methotrexate-Associated Lymphoproliferative Disorders Developed in the Oral Mucosa: A Case Series of 10 Patients and Literature Review
Kyoichi Obata, Tatsuo Okui, Sawako Ono, Koki Umemori, Shoji Ryumon, Kisho Ono, Mayumi Yao, Norie Yoshioka, Soichiro Ibaragi, Akira Sasaki
Diagnostics.2021; 11(8): 1375. CrossRef - Primary age‐related EBV‐associated effusion‐based lymphoma successfully treated with rituximab and thoracentesis
Justin J. Kuhlman, Muhamad Alhaj Moustafa, Alexander J. Tun, David M. Menke, Han W. Tun, Liuyan Jiang
Clinical Case Reports.2021;[Epub] CrossRef - Viral Manipulation of the Host Epigenome as a Driver of Virus-Induced Oncogenesis
Shimaa Hassan AbdelAziz Soliman, Arturo Orlacchio, Fabio Verginelli
Microorganisms.2021; 9(6): 1179. CrossRef - Spontaneous regression of chronic epstein –Barr virus infection-related lymphoproliferative disease
Bharti Kumari, Akshata Rao, ManickaSaravanan Subramanian, AparajitBallav Dey
Journal of the Indian Academy of Geriatrics.2021; 17(1): 40. CrossRef - The Pivotal Role of Viruses in the Pathogeny of Chronic Lymphocytic Leukemia: Monoclonal (Type 1) IgG K Cryoglobulinemia and Chronic Lymphocytic Leukemia Diagnosis in the Course of a Human Metapneumovirus Infection
Jérémy Barben, Alain Putot, Anca-Maria Mihai, Jérémie Vovelle, Patrick Manckoundia
Viruses.2021; 13(1): 115. CrossRef - B cells in multiple sclerosis — from targeted depletion to immune reconstitution therapies
Maria T. Cencioni, Miriam Mattoscio, Roberta Magliozzi, Amit Bar-Or, Paolo A. Muraro
Nature Reviews Neurology.2021; 17(7): 399. CrossRef - Development of Mast Cell and Eosinophil Hyperplasia and HLH/MAS-Like Disease in NSG-SGM3 Mice Receiving Human CD34+ Hematopoietic Stem Cells or Patient-Derived Leukemia Xenografts
Laura J. Janke, Denise M. Imai, Heather Tillman, Rosalinda Doty, Mark J. Hoenerhoff, Jiajie J. Xu, Zachary T. Freeman, Portia Allen, Natalie Wall Fowlkes, Ilaria Iacobucci, Kirsten Dickerson, Charles G. Mullighan, Peter Vogel, Jerold E. Rehg
Veterinary Pathology.2021; 58(1): 181. CrossRef - Viral coinfections in COVID‐19
Parisa S. Aghbash, Narges Eslami, Milad Shirvaliloo, Hossein B. Baghi
Journal of Medical Virology.2021; 93(9): 5310. CrossRef - Genetic predisposition to lymphomas: Overview of rare syndromes and inherited familial variants
Bartosz Szmyd, Wojciech Mlynarski, Agata Pastorczak
Mutation Research/Reviews in Mutation Research.2021; 788: 108386. CrossRef - Acute Epstein‐Barr virus associated haemophagocytosis in an Asian female: What is the diagnosis?
Soumya Ojha, Guiyi Ho, Cheryl X. Q. Lim, Siok B. Ng, Sanjay de Mel
American Journal of Hematology.2021; 96(11): 1541. CrossRef - Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases
Xinle Cui, Clifford M. Snapper
Frontiers in Immunology.2021;[Epub] CrossRef - Recent Advances in Diagnosis and Therapy of Angioimmunoblastic T Cell Lymphoma
Mostafa F. Mohammed Saleh, Ahmed Kotb, Ghada E. M. Abdallah, Ibrahim N. Muhsen, Riad El Fakih, Mahmoud Aljurf
Current Oncology.2021; 28(6): 5480. CrossRef - Intestinal ulcers as an initial finding in EBV-associated lymphoproliferative disorder
Sizhu Wang, Yinghuan Dai, Jie Zhang, Dalian Ou, Chunhui Ouyang, Fanggen Lu
Medicine.2020; 99(3): e18764. CrossRef - Microbes as Master Immunomodulators: Immunopathology, Cancer and Personalized Immunotherapies
Joana R. Lérias, Georgia Paraschoudi, Eric de Sousa, João Martins, Carolina Condeço, Nuno Figueiredo, Carlos Carvalho, Ernest Dodoo, Mireia Castillo-Martin, Antonio Beltrán, Dário Ligeiro, Martin Rao, Alimuddin Zumla, Markus Maeurer
Frontiers in Cell and Developmental Biology.2020;[Epub] CrossRef - Epstein Barr Virus-associated Pediatric Neoplasms
Mozhgan Hashemieh, Fariba Shirvani
Archives of Pediatric Infectious Diseases.2020;[Epub] CrossRef - Novel IRF8 and PD-L1 molecular aberrations in systemic EBV-positive T-cell lymphoma of childhood
Atif Saleem, Rohan Joshi, Li Lei, Lhara Lezama, Shyam S. Raghavan, Nastaran Neishaboori, Mohana Roy, Joe Schroers-Martin, Gregory W. Charville, Christian Kunder, Brent Tan, Beth A. Martin, Yasodha Natkunam
Human Pathology: Case Reports.2020; 19: 200356. CrossRef - Fatal SARS-CoV-2 coinfection in course of EBV-associated lymphoproliferative disease
Luca Roncati, Beatrice Lusenti, Vincenzo Nasillo, Antonio Manenti
Annals of Hematology.2020; 99(8): 1945. CrossRef - Epstein-Barr Virus and the Eye
Emmett T. Cunningham, Manfred Zierhut
Ocular Immunology and Inflammation.2020; 28(4): 533. CrossRef - An atypical systemic form of chronic active EBV infection
Neha Gupta, Adam Bagg
Leukemia & Lymphoma.2020; 61(12): 3030. CrossRef - A Shared TCR Bias toward an Immunogenic EBV Epitope Dominates in HLA-B*07:02–Expressing Individuals
Louise C. Rowntree, Thi H. O. Nguyen, Carine Farenc, Hanim Halim, Luca Hensen, Jamie Rossjohn, Tom C. Kotsimbos, Anthony W. Purcell, Katherine Kedzierska, Stephanie Gras, Nicole A. Mifsud
The Journal of Immunology.2020; 205(6): 1524. CrossRef - Chronic active Epstein–Barr virus infection manifesting as coronary artery aneurysm and uveitis
Haijuan Xiao, Bing Hu, Rongmu Luo, Huili Hu, Junmei Zhang, Weiying Kuang, Rui Zhang, Li Li, Gang Liu
Virology Journal.2020;[Epub] CrossRef - Epstein-Barr Virus (EBV)-induced B-cell Lymphoproliferative Disorder Mimicking the Recurrence of EBV-associated Hemophagocytic Lymphohistiocytosis
Yuki Yatsushiro, Takuro Nishikawa, Aki Saito, Yozo Nakazawa, Ken-Ichi Imadome, Shunsuke Nakagawa, Yuichi Kodama, Yasuhiro Okamoto, Hirokazu Kanegane, Yoshifumi Kawano
Journal of Pediatric Hematology/Oncology.2019; 41(1): e44. CrossRef - Epstein-Barr Virus (EBV)-Related Lymphoproliferative Disorders in Ataxia Telangiectasia: Does ATM Regulate EBV Life Cycle?
Moussab Tatfi, Olivier Hermine, Felipe Suarez
Frontiers in Immunology.2019;[Epub] CrossRef - The factors associated with the early diagnosis of nasal NK/T-cell lymphoma with prominent ocular symptoms and general nasal NKTL
Zhen zhen Hu, Ying Wang
American Journal of Otolaryngology.2019; 40(3): 353. CrossRef - Unusual lymphoid malignancy and treatment response in two children with Down syndrome
Ashley Geerlinks, Jennifer Keis, Bo Ngan, Amer Shammas, Reza Vali, Johann Hitzler
Pediatric Blood & Cancer.2019;[Epub] CrossRef - Extreme Peripheral Blood Plasmacytosis Mimicking Plasma Cell Leukemia as a Presenting Feature of Angioimmunoblastic T-Cell Lymphoma (AITL)
Kelsey Sokol, Saritha Kartan, William T. Johnson, Onder Alpdogan, Neda Nikbakht, Bradley M. Haverkos, Jerald Gong, Pierluigi Porcu
Frontiers in Oncology.2019;[Epub] CrossRef - High-Throughput Sequence Analysis of Peripheral T-Cell Lymphomas Indicates Subtype-Specific Viral Gene Expression Patterns and Immune Cell Microenvironments
Hani Nakhoul, Zhen Lin, Xia Wang, Claire Roberts, Yan Dong, Erik Flemington, Blossom Damania
mSphere.2019;[Epub] CrossRef - Quercetin Interrupts the Positive Feedback Loop Between STAT3 and IL-6, Promotes Autophagy, and Reduces ROS, Preventing EBV-Driven B Cell Immortalization
Granato, Gilardini Montani, Zompetta, Santarelli, Gonnella, Romeo, D’Orazi, Faggioni, Cirone
Biomolecules.2019; 9(9): 482. CrossRef - Diffuse Large B-Cell Lymphoma Arising within Ileal Neobladder: An Expanding Spectrum of Diffuse Large B-Cell Lymphoma Associated with Chronic Inflammation
Hyekyung Lee, Hyunbin Shin, Nae Yu Kim, Hyun Sik Park, Jinsung Park
Cancer Research and Treatment.2019; 51(4): 1666. CrossRef - EBV-associated lymphoproliferative disorder involving the gastrointestinal tract which mimic IBD in immunocompetent patients: case reports and literature review
Yanhua Zhou, Yanlin Zhang, Haiying Zhao, Xuan Cui, Yongqiu Wei, Yongdong Wu, Shutian Zhang, Ye Zong
International Journal of Colorectal Disease.2019; 34(11): 1989. CrossRef - Mechanistic Insights into Chemoresistance Mediated by Oncogenic Viruses in Lymphomas
Jungang Chen, Samantha Kendrick, Zhiqiang Qin
Viruses.2019; 11(12): 1161. CrossRef - Rapidly Fatal Encephalitis Associated with Atypical Lymphoid Proliferations of the Basal Ganglia Subsequent to Aneurysmal Subarachnoid Hemorrhage
Ayesha Kar, Evin L. Guilliams, Joshua A. Cuoco, Eric A. Marvin
Clinics and Practice.2019; 9(4): 1187. CrossRef - Clinicopathologic features of adult EBV-associated B-cell lymphoproliferative disease
Sonja Wörner, Hans-Konrad Mueller-Hermelink, Hans-Ullrich Voelker
Pathology - Research and Practice.2018; 214(2): 207. CrossRef - Primary Intestinal Epstein–Barr Virus-associated Natural Killer/T-cell Lymphoproliferative Disorder: A Disease Mimicking Inflammatory Bowel Disease
Zhujun Wang, Wenyan Zhang, Chengxin Luo, Min Zhu, Yu Zhen, Jingxi Mu, Yan Zhang, Renwei Hu, Yufang Wang, Zhonghui Wen, Qin Ouyang, Shuyuan Xiao, Hu Zhang
Journal of Crohn's and Colitis.2018; 12(8): 896. CrossRef - Downregulation of CD5 and dysregulated CD8+ T‐cell activation
Taizo Wada
Pediatrics International.2018; 60(9): 776. CrossRef - Chronic active Epstein-Barr virus infection of T-cell type, systemic form in an African migrant: case report and review of the literature on diagnostics standards and therapeutic options
Maxi Wass, Marcus Bauer, Roald Pfannes, Kerstin Lorenz, Andreas Odparlik, Lutz P Müller, Claudia Wickenhauser
BMC Cancer.2018;[Epub] CrossRef - Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy
Edit Porpaczy, Sabrina Tripolt, Andrea Hoelbl-Kovacic, Bettina Gisslinger, Zsuzsanna Bago-Horvath, Emilio Casanova-Hevia, Emmanuelle Clappier, Thomas Decker, Sabine Fajmann, Daniela A. Fux, Georg Greiner, Sinan Gueltekin, Gerwin Heller, Harald Herkner, Gr
Blood.2018; 132(7): 694. CrossRef - Gammaherpesviral infections in patients with immunological disorders
Anna Żuk-Wasek, Maciej Przybylski, Natalia Żeber, Grażyna Młynarczyk, Tomasz Dzieciątkowski
Postępy Mikrobiologii - Advancements of Microbiology.2018; 57(2): 145. CrossRef - COMPARATIVE ANALYSIS OF SEROLOGICAL MARKERS OF HERPES VIRUSES AND QUANTITATIVE IMMUNOGLOBULINOPATHIES IN PRIMARY PATIENTS WITH ANGIOIMMUNOBLASTIC T-CELL LYMPHOMA
N. G. Chernova, D. S. Tihomirov, N. P. Soboleva, S. A. Mariina, Y. V. Sidorova, M. N. Sinitsyna, V. N. Dvirnyk, S. M. Kulikov, T. A. Tupoleva, E. E. Zvonkov
Problems of Virology.2018; 63(4): 171. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-