 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 51(3); 2017 > Article
-
Brief Case Report
Cerebellar Liponeurocytoma: Relevant Clinical Cytogenetic Findings - Alexander Tucker, Kritsanapol Boon-Unge1, Nancy McLaughlin, Hassana Ibrahim2, Nagesh Rao3, Neil Martin, Richard Everson, Négar Khanlou1
-
Journal of Pathology and Translational Medicine 2017;51(3):335-340.
DOI: https://doi.org/10.4132/jptm.2016.07.24
Published online: October 16, 2016
Department of Neurosurgery, University of California-Los Angeles, Los Angeles, CA, USA
1Division of Neuropathology, Department of Pathology and Laboratory Medicine, University of California-Los Angeles, Los Angeles, CA, USA
2Division of Neuroradiology, Department of Radiological Sciences, University of California-Los Angeles, Los Angeles, CA, USA
3Division of Clinical and Molecular Cytogenetics, Department of Pathology and Laboratory Medicine, University of California-Los Angeles, Los Angeles, CA, USA
- Corresponding Author Alexander Tucker, MD Department of Neurosurgery, University of California-Los Angeles, Suite 562 5th Floor, Wasserman Building, 300 Stein Plaza, Los Angeles, CA 90095-6901, USA Tel: +1-310-794-7362, Fax: +1-310-267-2707, E-mail: atucker@mednet.ucla.edu
© 2017 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- A 41-year-old, right-handed woman without significant past medical or family history presented for evaluation of intermittent, positional headaches for 1 year. These symptoms progressed and became associated with nausea. The patient denied any postural instability, tremor, changes in gait, vision changes, dysphonia, weakness, or sensory disturbance. There was no associated neck or back pain.
- Examination at presentation revealed that the patient was awake, alert, and fully oriented. Visual fields were full to confrontation and fundoscopic examination showed no papilledema. The patient had normal facial sensation and facial movement. Tongue and palate were along the midline. Bilateral finger-to-nose testing was normal. Strength was full and no pronator drift was detected. Sensation was intact to light touch throughout. No hyperreflexia was noted nor were there signs of lower extremity clonus. An equivocal Babinski sign was noted on the right side, but toes were downgoing on the left. Her gait appeared smooth and Romberg sign was negative.
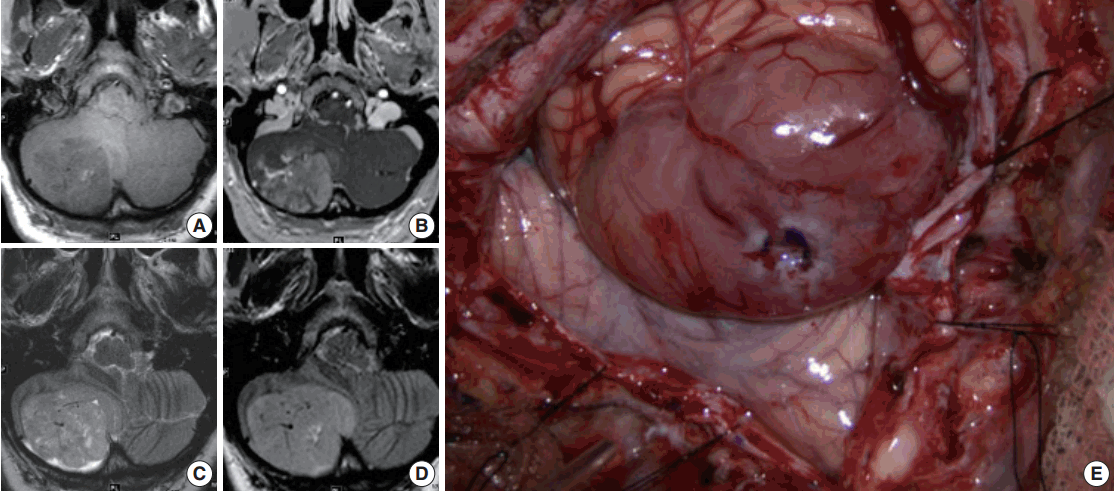
- Magnetic resonance imaging (MRI) of the brain with and without contrast was obtained and showed a 4.5 × 5.2 ×3 .6 cm (anterior-posterior × transverse × cranial-caudal) heterogeneously enhancing mass in the right cerebellar hemisphere. The lesion was hypointense on T1 and hyperintense on T2, and it had minimal surrounding edema (Fig. 1A–D). The presence of flow voids on T2 imaging indicated possible high vascularity. It was not entirely clear if the tumor was intra- or extra-axial. Moderate mass effect was evident, resulting in tonsillar herniation of approximately 1.6 cm into the foramen magnum and compression of the fourth ventricle. Resultant obstructive hydrocephalus was mild, and transependymal transudation was noted on T2 imaging. Complete spine MRI did not show any abnormality.
- Suboccipital craniotomy was performed and microsurgical total resection of the tumor was achieved. Grossly, the tumor appeared to be intra-axial with an exophytic component and it was red/yellow-colored, not hemorrhagic, and well-circumscribed. It originated within the medial aspect of the right cerebellar hemisphere in the region of the vermis underlying the arachnoid (Fig. 1E). It did not involve the dura. Superficially, the tumor had a clearly defined margin with a clean plane between the lesion and the adjacent cerebellar tissue. In the deep aspect of the tumor, however, the interface between the tumor and healthy parenchyma was not as sharply defined.
- Histopathology and molecular studies
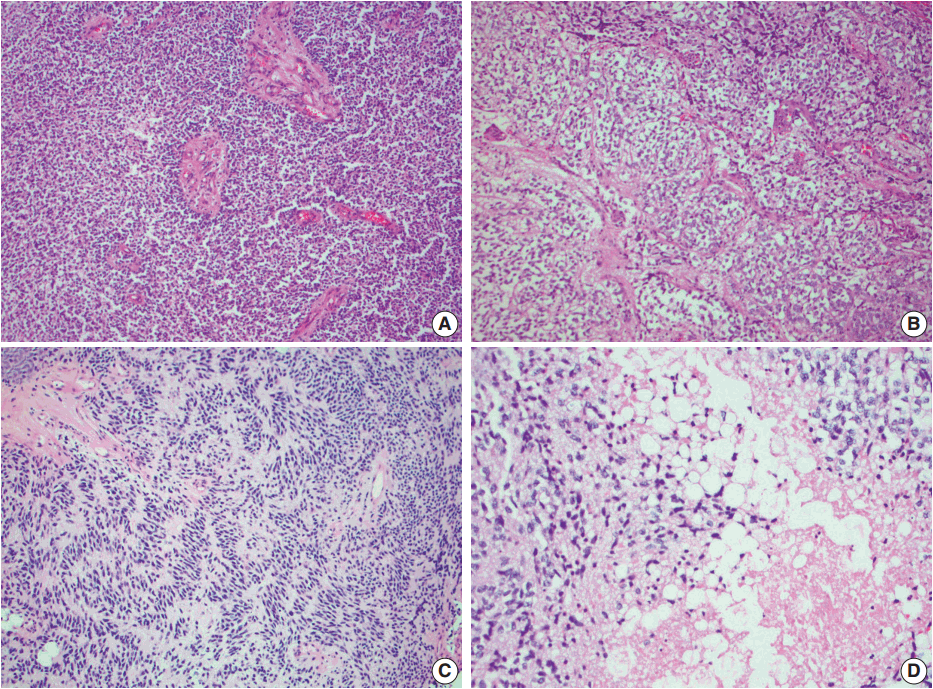
- Histologic examination of the permanent sections showed a moderately cellular biphasic neoplasm. Areas of tumor cells were characterized by round to elongated nuclei with scant cytoplasm arranged in sheets (Fig. 2A). Foci of oligodendroglia-like tumor cells with central round nuclei and perinuclear halos were arranged in nests, separated by thin branching vessels (Fig. 2B). In other areas, nuclear palisading, perivascular rosetting, and rare Homer-Wright rosettes were seen (Fig. 2C). Nuclear pleomorphism and atypia were minimal. Clusters of lipidized tumor cells reminiscent of mature adipocytes were easily identified within the tumor (Fig. 2D). Mitoses were inconspicuous. Rare microfoci of necrosis were seen whereas pseudopalisading necrosis was absent. Prominent vascular channels with hyalinization and occasional complex vessels were noted within the tumor. Classic microvascular endothelial hyperplasia was not identified.
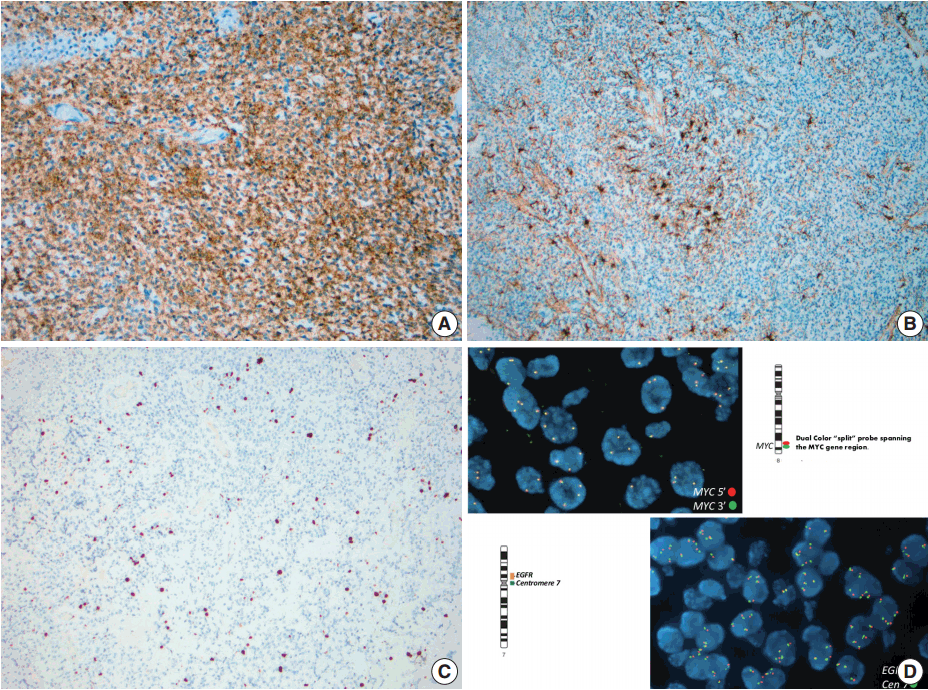
- Immunohistochemistry using antibodies to glial fibrillary acidic protein (GFAP), a glial marker, and synaptophysin, a neuronal marker, were performed. Neoplastic cells showed diffuse and strong immunoreactivity with synaptophysin (Fig. 3A), but failed to react with GFAP. Staining for GFAP and CD34 highlighted background reactive gliosis and blood vessels, respectively (Fig. 3B). Inhibin, CD99, and neurofilament immunoreactions were negative in tumor cells. The proliferative rate based on Ki-67 nuclear labeling index was estimated at 3%–5% (Fig. 3C). Although overall morphologic and immunohistochemical profile did not favor a high grade tumor, select molecular studies were performed.
- Fluorescent in-situ hybridization (FISH) studies showed no evidence of 1p/19q co-deletion, N-MYC or epidermal growth factor receptor (EGFR) amplification, MYC or EWSR1 rearrangements, or PTEN deletion. However, these studies detected three to six copies each of chromosomes 1, 2, 7, 8q, 10, 19, and 22q in 50%–80% ([151–247]/300) of nuclei examined (Fig. 3D).
CASE REPORT
- The case described here highlights the main clinical, radiographic and pathologic features of cLNC. With its histologic similarity to many other neoplasms, surgical pathologists must include cLNC in the differential diagnoses of lesions of the posterior fossa with a clear cell morphology (oligodendroglioma, hemangioblastoma, and clear cell ependymoma), neuroectodermal tumors (medulloblastoma), and tumors with possible lipidized component (glioblastoma and meningioma). Without a clear diagnostic rubric, most histologic and molecular studies aim to exclude tumors of high-grade potential, rendering the diagnosis of cLNC as a diagnosis of exclusion. The importance of accurate diagnosis cannot be overstated, as it used to guide further therapeutic intervention and determination of the patient’s prognosis.
- Histologically, cLNC appears as monotonous sheets of clear cells or oligodendroglia-like cells, as well as characteristic lipidized cells. Biphasic patterns of dense cellularity alternating with clear cell areas, such as in this case, have also been reported [3]. Nuclear palisading, true rosette formation and perivascular arrangements were also noted in our case; however, this is not typical. Areas of high cellularity and rosette formation may prompt some pathologists to consider the diagnosis of glioblastoma or ependymoma; however, unlike these entities, cLNC is rarely mitotically active. Features such as necrosis and vascular endothelial hyperplasia, when present, are not correlated with malignant transformation or rapid regrowth and have no effect on overall prognosis. Conversely, there are a few reports describing aggressive clinical tumor behavior in the absence of the classic high-grade features, although this evidence is limited [5].
- The most common alternative diagnosis considered by pathologists reviewing cases of cLNC is medulloblastoma. Medulloblastoma is the most prevalent primary malignancy of the posterior fossa, seen most commonly in children and young adults. In contrast, cLNC is typically diagnosed in adults. Histologically, medulloblastoma has a more primitive or embryonal appearance than the neurocytic morphology observed in the cLNC. Similarly, anaplasia, nuclear crowding, and high proliferation rates are more common in medulloblastoma than in cLNC. In addition, clear cell morphology and lipidized cells are not typical findings in medulloblastoma, but are seen in cLNC.
- Other tumors that are considered part of the differential diagnosis for cLNC include oligodendroglioma and clear cell ependymoma. In general, the diagnosis of oligodendroglioma in the cerebellum should be approached cautiously and cLNC should always be in included in its differential diagnosis. Despite clear cell morphology, oligodendroglioma lacks the extensive neuronal differentiation or lipidization that is found in cLNC. Similarly, clear cell ependymoma is generally strongly immunoreactive to GFAP, lacks lipid-laden cells, and histologically presents with higher-grade nuclear features.
- When studied with immunohistochemistry, cLNC is immunoreactive with the most commonly used neuronal markers (i.e., neuron-specific enolase, synaptophysin, Neu-N, chromogranin A, and MAP-2). Glial differentiation and GFAP expression when present are limited. Cases with skeletal muscle differentiation, albeit uncommon, may show desmin expression [6]. The Ki-67 proliferation index for cLNC is generally significantly lower than that for medulloblastoma and other high grade mimics, typically < 5% [7]. In many cases a low mitotic rate and low Ki-67 proliferation index can be used to exclude medulloblastoma as a likely consideration when histological diagnosis is less clear.
- Although current literature tends to use histologic and molecular markers for neurocytomas to classify cLNC, recent studies question this practice and suggest that cLNC may have an alternative genetic pathogenesis. In central neurocytoma, polysomic gain of chromosome 7 is found frequently and isolated loss of 1p and 19q have also been reported [8]. In extraventricular neurocytomas, dual 1p/19q loss and particularly t(1;19)(q10; p10), is observed and has been associated with atypical histologic features and increased mitotic activity [9]. It should be noted that dual 1p/19q loss is considered the genetic hallmark of oligodendroglioma and is not seen in central neurocytoma. In contrast to central and extraventricular neurocytoma, there are no detailed cytogenetic reports of cLNC tumors.
- In the case presented here, there was no evidence of 1p/19q co-deletion, N-MYC or EGFR amplification, MYC or EWSR1 rearrangements, or PTEN deletion. Cytogenetic studies detected three to six copies each of chromosomes 1, 2, 7, 8q, 10, 19, and 22q. In addition to preserved MYC status, detailed cluster DNA analyses was used to distinguish cLNC from medulloblastoma. Typically cLNC lacks isochromosome 17 or mutations of the beta-catenin (CTNNB1), PTCH or adenomatous polyposis coli (APC) genes which are often found in medulloblastoma [2,10]. Unlike astrocytic tumors, IDH1 mutation is not commonly present in cLNC and there was no IDH1 mutation in codon 132 in the case presented here. TP53 mutations are seen in 20% of the cLNC cases, although not in the case described above, which is a higher rate than in medulloblastoma and usually not found in central neurocytoma [10]. Recent data show the upregulation of FABP4, a gene primarily expressed in adipocytes and macrophages. Unfortunately most of these molecular markers are not available to clinical testing. The specificity or prognostic significance of these finding remains to be determined, although it appears likely that cLNC is a genetically distinct entity from other forms of neurocytoma. We recommend that role of FISH studies in the diagnosis of cLNC is to test for clinically relevant genetic abnormalities known to be associated with histologic mimics of cLNC as their absence could be of tremendous diagnostic and prognostic value (Table 1).
- According to the WHO classification, cLNC is considered a low-grade lesion; however, long-term follow-up of patients diagnosed with this tumor is imperative. Recent research suggests that the recurrence rate for cLNC may be much higher than previously believed-as high as 40% within 12 years for patients treated surgically [2]. Currently, the standard of care for treating these tumors involves surgical resection and serial neuroimaging. Although evidence is limited, some clinicians advocate adjuvant radiotherapy in cases of subtotal resection or recurrence if reoperation is not possible [5]. Like most primary intracranial neoplasms, extra cranial metastasis is rare and only one case of distant metastases has been reported in the literature. There are currently no accepted histologic features of cLNC that can be relied on to indicate a more aggressive nature of increased likelihood of recurrence. Atypical features including high proliferative index, necrosis, high mitotic activity, and prominent vascular hyperplasia may warrant a close clinical follow-up, although these findings have not yet been correlated with clinical outcome. It remains to be elucidated whether these atypical features are truly predictive of a more malignant biologic behavior since some tumors recur in the absence of atypical histopathology.
- This case report is the first complete description of the clinical, radiographic, histologic, immunohistochemical and genetic findings of cLNC. Although no one immunohistochemical marker, genetic profile or histologic feature distinguishes cLNC, a constellation of findings from multiple modalities can be used to correctly diagnosis this lesion. Further study of cLNC will be required to better understand its pathophysiology, identify a unique diagnostic indicator and perhaps find a cure for this rare brain tumor.
DISCUSSION
- 1. Bechtel JT, Patton JM, Takei Y. Mixed mesenchymal and neuroectodermal tumor of the cerebellum. Acta Neuropathol 1978; 41: 261-3. ArticlePubMedPDF
- 2. Nishimoto T, Kaya B. Cerebellar liponeurocytoma. Arch Pathol Lab Med 2012; 136: 965-9. ArticlePubMedPDF
- 3. George DH, Scheithauer BW. Central liponeurocytoma. Am J Surg Pathol 2001; 25: 1551-5. ArticlePubMed
- 4. Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007; 114: 97-109. ArticlePubMedPMCPDF
- 5. Jenkinson MD, Bosma JJ, Du Plessis D, et al. Cerebellar liponeurocytoma with an unusually aggressive clinical course: case report. Neurosurgery 2003; 53: 1425-7. ArticlePubMedPDF
- 6. Gonzalez-Campora R, Weller RO. Lipidized mature neuroectodermal tumour of the cerebellum with myoid differentiation. Neuropathol Appl Neurobiol 1998; 24: 397-402. ArticlePubMedPDF
- 7. Oudrhiri MY, Raouzi N, El Kacemi I, et al. Understanding cerebellar liponeurocytomas: case report and literature review. Case Rep Neurol Med 2014; 2014: 186826.ArticlePubMedPMCPDF
- 8. Taruscio D, Danesi R, Montaldi A, Cerasoli S, Cenacchi G, Giangaspero F. Nonrandom gain of chromosome 7 in central neurocytoma: a chromosomal analysis and fluorescence in situ hybridization study. Virchows Arch 1997; 430: 47-51. ArticlePubMedPDF
- 9. Rodriguez FJ, Mota RA, Scheithauer BW, et al. Interphase cytogenetics for 1p19q and t(1;19)(q10;p10) may distinguish prognostically relevant subgroups in extraventricular neurocytoma. Brain Pathol 2009; 19: 623-9. ArticlePubMed
- 10. Horstmann S, Perry A, Reifenberger G, et al. Genetic and expression profiles of cerebellar liponeurocytomas. Brain Pathol 2004; 14: 281-9. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- CEREBELLAR LIPONEUROCYTOMA IN THE LAST DECADE: A COMPREHENSIVE CASE REVIEW
Oskar Sienkiel, Julia Czerwik, Marcin Narloch, Jan Tomczyk, Mateusz Jasiński, Aleksandra Żywicka, Michał Szalach, Kamila Mozga, Mathias Spitaleri, Dawid Sewruk, Karol Kanon
International Journal of Innovative Technologies in Social Science.2025;[Epub] CrossRef - Cerebellar liponeurocytoma: Rare posterior fossa tumor
Ismail Chaouche, Nizar EL Bouardi, Btissam Benabderrazik, Meriem Haloua, Moulay youssef alaoui Lamrani, Maryam Boubbou, Mustapha Maaroufi, Badreeddine Alami
Radiology Case Reports.2024; 19(8): 3382. CrossRef - Cerebellar Liponeurocytoma: Publication Trends, Scientometrics Analysis, and Critical Review
Rabia Ali, Sulaman Durrani, Karim Rizwan Nathani, Ryan Jarrah, Mohamad Bydon
World Neurosurgery.2023; 171: e137. CrossRef - Surgical management and clinical outcomes of cerebellar liponeurocytomas—a report of seven cases and a pooled analysis of individual patient data
Pengcheng Zuo, Tao Sun, Guocan Gu, Xiaoou Li, Zhuang Jiang, Changcun Pan, Cheng Xu, Zhen Wu, Junting Zhang, Liwei Zhang
Neurosurgical Review.2022; 45(2): 1747. CrossRef - Cerebellar liponeurocytoma: clinical, histopathological and molecular features of a series of three cases, including one recurrent tumor
Giuseppe Broggi, Elena Tirrò, Hiba Alzoubi, Antonietta Arcella, Francesca Gianno, Manila Antonelli, Simone Minasi, Paolo Vigneri, Francesco Certo, Roberto Altieri, Giuseppe Maria Vincenzo Barbagallo, Evelina Miele, Rosario Caltabiano, Felice Giangaspero
Neuropathology.2022; 42(3): 169. CrossRef - Cerebellar Liponeurocytoma Mimicking Medulloblastoma: Case Report of a Childhood and Literature Review
Changhui Dong, Yining Jiang, Liyan Zhao, Yubo Wang, Yang Bai, Ying Sun, Yunqian Li
Frontiers in Oncology.2021;[Epub] CrossRef - Cerebellar liponeurocytoma - molecular signature of a rare entity and the importance of an accurate diagnosis
Thomas Linsenmann, Camelia M. Monoranu, Balint Alkonyi, Thomas Westermaier, Carsten Hagemann, Almuth F. Kessler, Ralf-Ingo Ernestus, Mario Löhr
Interdisciplinary Neurosurgery.2019; 16: 7. CrossRef - Cerebellar liponeurocytoma – a rare entity: a case report
Oliver Gembruch, Andreas Junker, Yahya Ahmadipour, Ulrich Sure, Elias Lemonas
Journal of Medical Case Reports.2018;[Epub] CrossRef - Liponeurocytoma: Systematic Review of a Rare Entity
Oliver Gembruch, Andreas Junker, Christoph Mönninghoff, Yahya Ahmadipour, Marvin Darkwah Oppong, Ulrich Sure, Nicolai El Hindy, Elias Lemonas
World Neurosurgery.2018; 120: 214. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
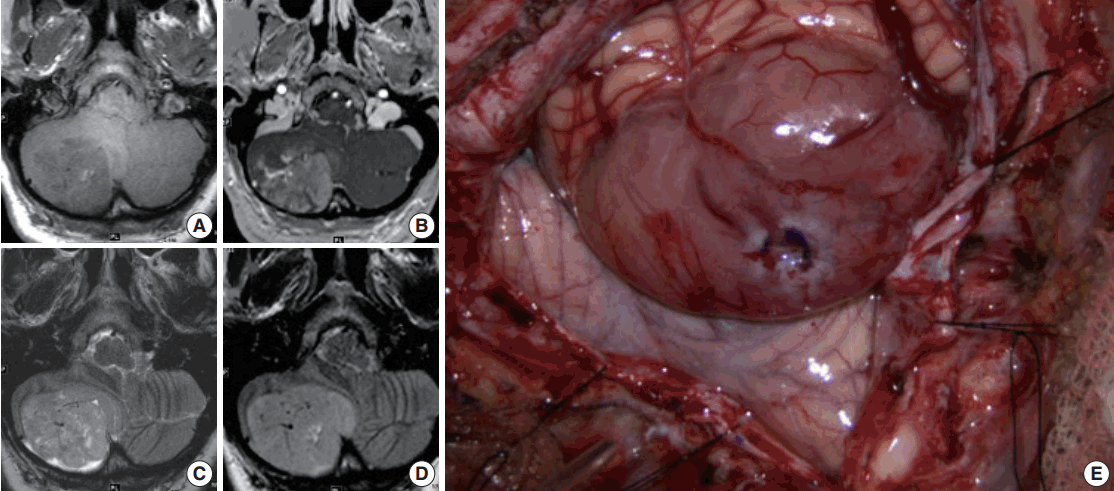
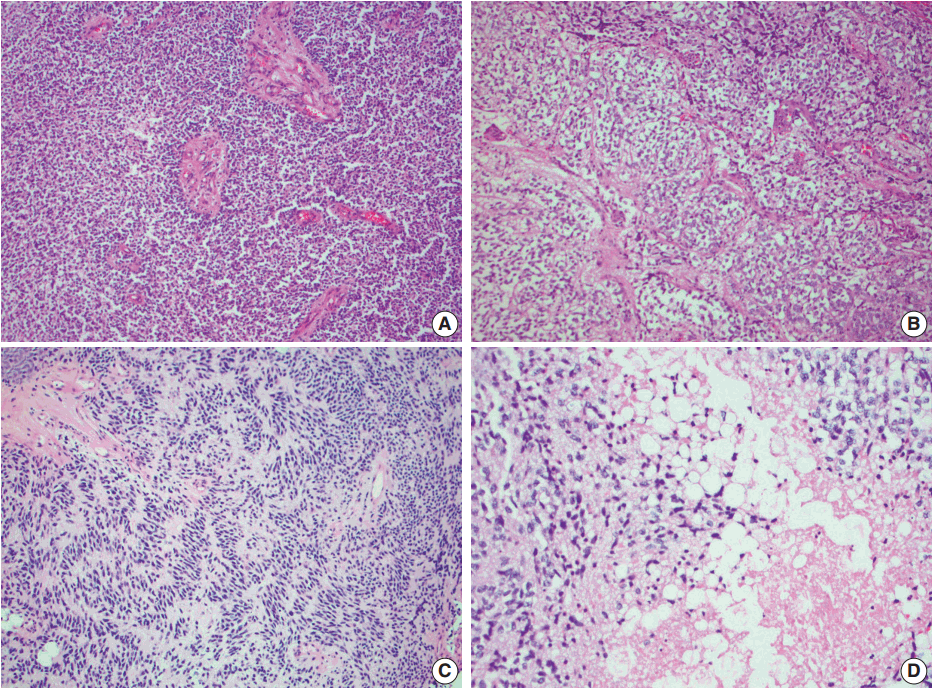
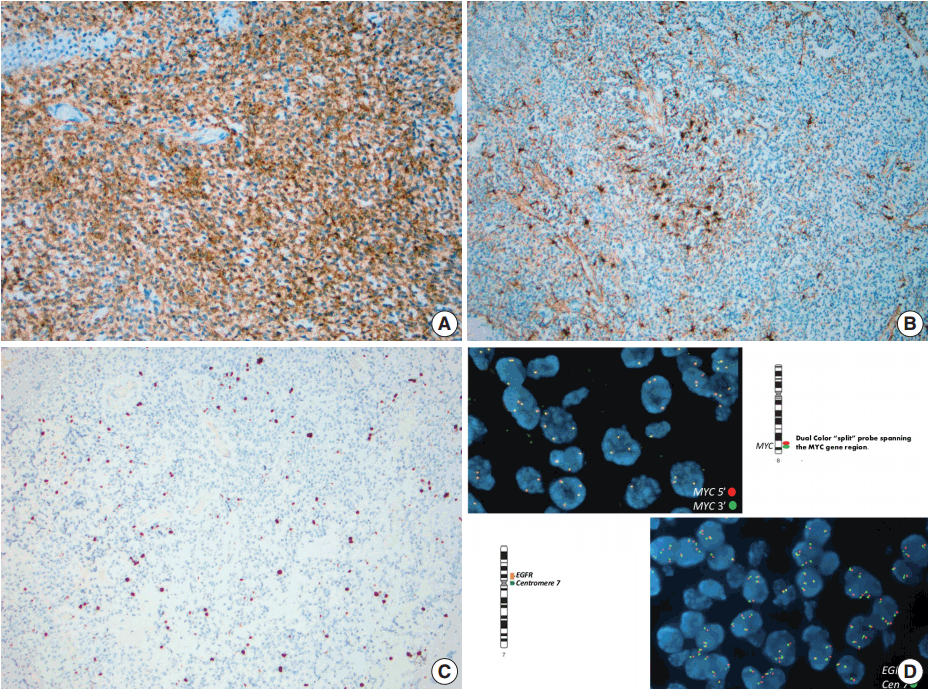



Fig. 1.
Fig. 2.
Fig. 3.
| Cerebellar liponeurocytoma | Central neurocytoma | Clear cell ependymoma | Oligodendroglioma | Medulloblastoma | |
|---|---|---|---|---|---|
| Age of diagnosis (yr) | 50–60 | 20–40 | 1-20 | 30–50 | 5–30 |
| WHO grade | II | II | II | II or III | IV |
| Histology | Neurocytic clear cells in sheets with lipidized cells | Clear cells, modest cytoplasm, salt and pepper chromatin | Clear cells, round nuclei, perinuclear halos, perivascular rosettes | Clear cells, dark nuclei, “chicken wire” vasculature | Highly cellular, small, round, primitive/embryonal cells |
| High grade features | Uncommon | Uncommon | Common | Common | Common |
| IHC hallmark | Synaptophysin + | Synaptophysin + | GFAP + | IDH-1 (R132H) +/- | Synaptophysin ++ |
| GFAP +/- | GFAP - | EMA + (dot-like) | Olig2 + | NSE ++ | |
| Ki-67 < 5% | Ki-67 < 5% | Vimentin ++ | Synaptophysin +/- | Vimentin ++ | |
| Ki-67 < 5% | Ki-67 variable | Ki-67 high (>20%) | |||
| Genetic hallmark | Preserved MYC status, no 1p/19q loss, no IDH1 mutation, possible TP53 mutation | Gain of chromosome 7 | Loss of chromosome 22 | Dual 1p/19q loss | Isochromosome 17q, MYC rearrangement |
cLNC, cerebellar liponeurocytoma; WHO, World Health Organization; IHC, immunohistochemisty; GFAP, glial fibrillary acidic protein; EMA, epithelial membrane antigen; IDH-1, isocitrate dehydrogenase 1; NSE, neuron-specific enolase.

