 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 51(3); 2017 > Article
-
Review
Molecular Testing of Brain Tumor - Sung-Hye Park,1,2, Jaekyung Won1, Seong-Ik Kim1, Yujin Lee1, Chul-Kee Park3, Seung-Ki Kim3, Seung-Hong Choi4
-
Journal of Pathology and Translational Medicine 2017;51(3):205-223.
DOI: https://doi.org/10.4132/jptm.2017.03.08
Published online: May 12, 2017
1Department of Pathology, Seoul National University, College of Medicine, Seoul, Korea
2Neurosicence Institute, Seoul National University, College of Medicine, Seoul, Korea
3Department of Neurosurgery, Seoul National University, College of Medicine, Seoul, Korea
4Department of Radiology, Seoul National University, College of Medicine, Seoul, Korea
- Corresponding Author Sung-Hye Park, MD, PhD Department of Pathology, Seoul National University Hospital, Seoul National University College of Medicine, 103 Daehak-ro, Jongno-gu, Seoul 03080, Korea Tel: +82-2-2072-3090 Fax: +82-2-743-5530 E-mail: 'shparknp@snu.ac.kr'
© 2017 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- The World Health Organization (WHO) classification of central nervous system (CNS) tumors was revised in 2016 with a basis on the integrated diagnosis of molecular genetics. We herein provide the guidelines for using molecular genetic tests in routine pathological practice for an accurate diagnosis and appropriate management. While astrocytomas and IDH-mutant (secondary) glioblastomas are characterized by the mutational status of IDH, TP53, and ATRX, oligodendrogliomas have a 1p/19q codeletion and mutations in IDH, CIC, FUBP1, and the promoter region of telomerase reverse transcriptase (TERTp). IDH-wildtype (primary) glioblastomas typically lack mutations in IDH, but are characterized by copy number variations of EGFR, PTEN, CDKN2A/B, PDGFRA, and NF1 as well as mutations of TERTp. High-grade pediatric gliomas differ from those of adult gliomas, consisting of mutations in H3F3A, ATRX, and DAXX, but not in IDH genes. In contrast, well-circumscribed low-grade neuroepithelial tumors in children, such as pilocytic astrocytoma, pleomorphic xanthoastrocytoma, and ganglioglioma, often have mutations or activating rearrangements in the BRAF, FGFR1, and MYB genes. Other CNS tumors, such as ependymomas, neuronal and glioneuronal tumors, embryonal tumors, meningothelial, and other mesenchymal tumors have important genetic alterations, many of which are diagnostic, prognostic, and predictive markers and therapeutic targets. Therefore, the neuropathological evaluation of brain tumors is increasingly dependent on molecular genetic tests for proper classification, prediction of biological behavior and patient management. Identifying these gene abnormalities requires cost-effective and high-throughput testing, such as next-generation sequencing. Overall, this paper reviews the global guidelines and diagnostic algorithms for molecular genetic testing of brain tumors.
- The indications and methods of each genetic study common to brain tumors are summarized in Tables 1–6. Integration of morphological criteria and genetic alterations is the guideline for an accurate diagnosis, risk classification, and development of therapeutic strategies.
- Mutations in IDH1/2
- The discovery of mutations affecting the enzymatic function of IDH in gliomas provided a fundamentally new insight into the biology of gliomagenesis and triggered molecular classification of gliomas by a somatic mutation. IDH catalyzes the oxidative decarboxylation of isocitrate to produce α-ketoglutarate (α-KG) and CO2, but mutant IDH1/2 has a preferential affinity for α-KG instead of isocitrate outside of the citric acid cycle, thus leading to the production and accumulation of the oncometabolite 2-hydroxyglutarate (2HG) [67-70]. Dang et al. [68] hypothesized that 2HG induces redox stress due to damage to the respiratory chain, which subsequently promotes the mutagenesis and development of gliomas. The α-KG–dependent prolyl hydroxylases modulate HIF1α levels and changes in the HIF1α downstream pathway, leading to an increase in reactive oxygen species levels and potentially contributing to the risk of cancer [71].
- The glioma-CpG island methylator phenotype (G-CIMP) subset is distinctively and invariably found in the gliomas with mutant IDH, which is the proneural transcriptional group [72]. 2HG, the oncometabolite produced by mutant IDH1/2, inhibits α-KG–dependent dioxygenases including histone demethylases and the Ten-Eleven Translocation (TET) family of histone 5-methylcytosine hydroxylase, which directly induce the hypermethylated state [73]. The G-CIMP subgroup of GBMs is common in younger patients and has a longer lifespan [13,17,30,67,74-76].
- All IDH1/2 mutations are common in 70%–80% of type II and III infiltrating gliomas and are found in 100% of oligoden-drogliomas and IDH-mutant GBMs [67].
- TP53 is a typical tumor suppressor gene located in 17p13.1, which encodes the nuclear protein p53. The p53 protein responds to diverse cellular stresses to regulate the expression of its target genes, thereby inducing cell cycle arrest, apoptosis, senescence, DNA repair, or metabolic changes. Mutated TP53 genes and overexpressed abnormal p53 protein, which has a longer half-life than wild type p53, are associated with a variety of human cancers, including Li-Fraumeni syndrome and many hereditary gliomas. As mentioned in the introduction, the p53 signaling pathway is one of the major abrogated pathways of astrocytic tumors including GBMs. WHO class II and III astrocytic tumors show high levels of TP53 mutations (94%) and/or p53 overexpression (Tables 3, 4) [3]. However, these alterations are rare in oligodendrogliomas, well-circumscribed astrocytic tumors including pilocytic astrocytomas, pleomorphic xanthoastrocytomas, and subependymal giant cell astrocytomas, ependymomas, and embryonal tumors such as medulloblastoma, except for the SHH-type, p53-activated subgroup.
- ATRX is an X-linked gene of α-thalassemia and mental retardation syndrome
- ATRX is located in Xq21.1, encodes a 280-kDa nuclear protein, and has been shown to be involved in a wide range of cellular functions such as DNA recombination, repair, and transcription regulation [82]. It binds strongly within the promyelocytic leukemia body of the nucleus. When ATRX interacts with DAXX, this complex functions as a histone chaperone complex for the deposition of histone variant H3.3 into heterochromatic repeats including pericentric, telomeric, and ribosomal DNA repeat sequences [83]. Functional mutations of this gene have been associated with sister chromatid clumps and defects, abnormal DNA methylation, and the maintenance of telomerase-independent telomeres, resulting in an alternative lengthening of telomeres (ALT). Decreased nuclear expression of ATRX and ATRX mutations are observed in grade 2 and 3 astrocytic tumors including IDH mutant gliomas (86%), IDH-mutant GBMs (85%), and pediatric high grade gliomas such as diffuse midline gliomas (DMGs) (Figs. 1, 2) [3].
- Histone H3F3A (K27M, K36, and G34) mutations
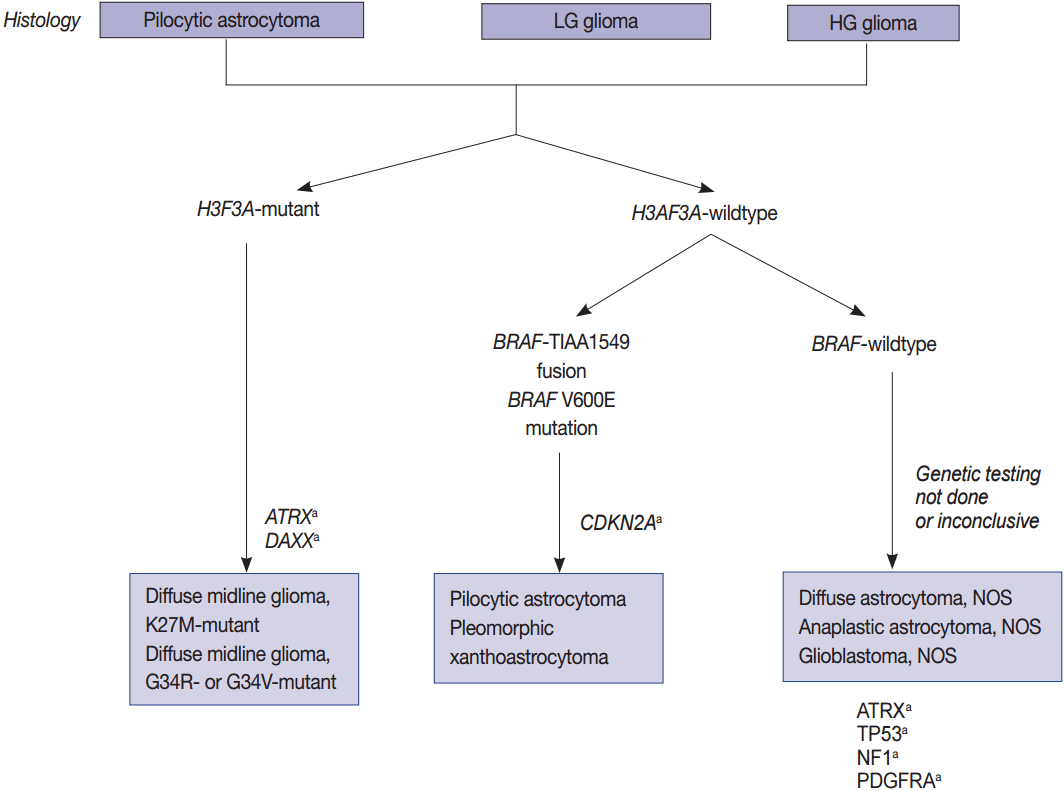
- Histone protein H3.1 and H3.2 are expressed only in the S phase and are integrated into the chromatin by chaperone CAF1 during DNA replication only, but H3.3 is expressed at all stages of the cell cycle and can be integrated into the chromatin independently of DNA replication. H3.3 is associated with active chromatin such as H3K4 trimethylation, which promotes open chromatin structure for the binding of co-factors and transcription factors to activate transcription [85]. Recurrent mutations in H3F3A, which encodes the replication-independent histone 3 variant H3.3, were first detected in the pediatric diffuse infiltrating pontine glioma (DIPG) by a comprehensive WES analysis. DIPG was later renamed as DMG and is considered a H3 K27M mutation in the new 2016 WHO classification (Fig. 2) [7,23]. Eighty percent of pontine gliomas (DIPG) and 70% of other loci DMGs contain a mutation in one allele of the H3F3A gene, which is a mutation on the histone tail (K27M, K36, G34R/G34V), an important post-translational modification factor [25].
- The histone H3F3A mutant gliomas have a poor prognosis regardless of histopathological grade. K27M-H3.3 mutations occur mainy in young patients (median age, 11 years) and G34R/V-H3.3 mutations occur in older children and young adults (median age, 20 years [22]. All of the cases with G34-H3.3 mutations are in pediatric GBMs (13/13), and especially associated with mutations in ATRX and DAXX [22]. Loss of ATRX is associated with the ALT phenotype; thus, ALT often coexists with mutations in ATRX/H3F3A/TP53 [22].
- Codeletion of chromosome 1p/19q and mutations in CIC and FUBP1
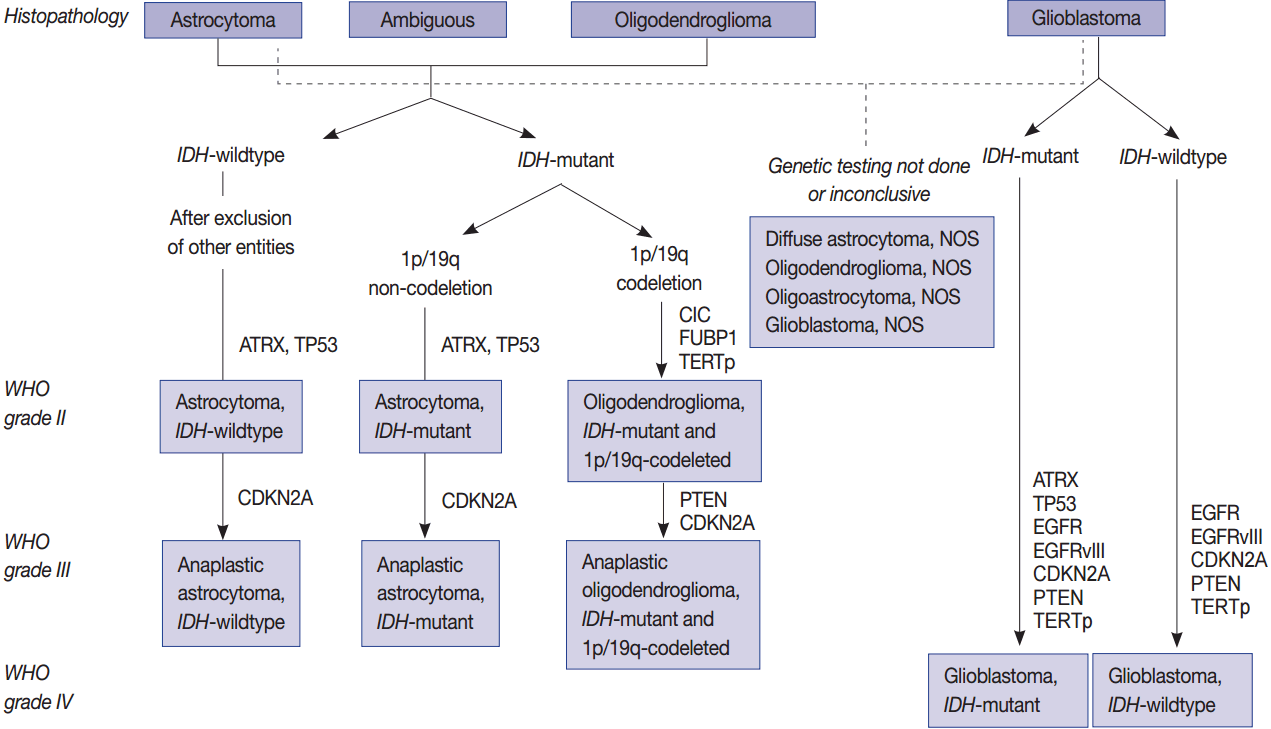
- Simultaneous deletion of chromosome 1p/19q is a characteristic and early genetic event in oligodendroglial tumors, which is associated with better prognosis and is also a good indicator of the patient response to specific combination chemotherapy of procarbazine, lomustine, and vincristine (PCV). Recurrent mutations in CIC and FUBP1 were found in 46%–53% and 15%–24% of oligodendrogliomas, respectively, in addition to a codeletion in 1p/19q and mutations in IDH1 or IDH2 (Fig. 1) [86-88].
- FUBP1 is located in 1p31.1, and mutations in FUBP1 are predicted to disrupt FUBP1 protein function (Table 1). FUBP1 acts as an RNA binding protein and alterations of its normal function can lead to tumorigenesis, which has been suggested to act either as a protooncogene or a tumor suppressor gene depending on the tumor type [36,87]. Through the c-Myc pathway, FUBP1 promotes cell migration and protects cells from apoptosis.
- The CIC gene on chromosome 19q13.2 represses genes induced by RTK pathway activation [89]. Although the function of human CIC protein is not known, a recent study in cultured cells demonstrated that CIC acts together with IDH1 to regulate citrate levels in the cytoplasm. CIC mutations promote the accumulation of 2HG and reduce clonogenicity in the setting of IDH1 mutations. Loss of 19q in oligodendroglial tumors unmasks mutations in the CIC gene [87,90]. In this regard, the co-existence of IDH1 with CIC or FUBP1 mutations may partially explain the slower tumor growth and longer survival of oligodendrogliomas with mutations in IDH1 and a codeletion of 1p/19q when compared to other diffuse gliomas [90]. The overall survival rate of patients with oligodendrogliomas with CIC mutations was lower than that of patients without CIC mutations, and the FUBP1 mutation was significantly associated with unfavorable progression-free survival [15]. However, oligodendrogliomas cannot be diagnosed with CIC or FUBP1 mutations only [15].
- EGFR amplification and EGFRvIII truncation mutations
- EGFR, also called Erb1 or HER1, is located on chromosome 7q12 and acts as an ErbB family receptor tyrosine kinase. EGFR amplification is closely related to EGFR overexpression. EGFRvIII-positive tumor cells exhibit particularly high levels of mTOR signal and exhibit the most proliferative and aggressive phenotype similar to GBMs (Fig. 1). The EGFRvIII mutation is a 801 bp frame deletion from exons 2 to 7 of the EGFR gene, which is associated with EGFR amplification and the response to antibody therapy as well as poor prognosis [93]. In addition to EGFR amplification, EGFRvIII-positive GBM cell lines are sensitively suppressed by first-generation EGFR inhibitors (erlotinib, gefitinib, vandetanib) and second-generation EGFR inhibitors (NT113) as compared to GBM cell lines in which PTEN tumor suppressor genes are lost in vitro [94].
- EGFR immunohistochemistry is usually uniform in tumors expressing high levels of the protein, however, EGFRvIII staining is heterogeneous and usually present in only a subset of tumor cells. Therefore, representative sampling of tumor tissue is important to avoid false-negative results [56,57].
- Unlike EGFR point mutations, EGFR fusions with SEPT14, PSPH, or SEC61G has also been shown to play a role in GBMs, offering a unique opportunity to investigate fused oncogene dependencies [94].
- Deletions in CDKN2A/2B
- CDKN2A/2B is a tumor suppressor gene located on chromosome 9p21.3, because it encodes a cyclin-dependent kinase inhibitor (p16) and is a cell cycle regulator of the Rb pathway. In 60% of GBMs and 11% of low-grade gliomas including oligodendroglioma, pleomorphic xanthoastrocytoma, and pilocytic astrocytoma, the loss or inactivation of p16 protein as a result of a homozygous deletion or promoter methylation of CDKN2A/2B is observed (Fig. 1) [55]. It is also one of the more commonly altered genes in PXA and high grade gliomas in both adults and children (Fig. 2) [35,95]. After adjusting for the IDH mutational status, sex, and age, CDKN2A deletions were strongly associated with poorer overall survival in astrocytomas but not in oligodendrogliomas [18].
- BRAF mutations and BRAF-KIAA1549 fusions
- BRAF V600E mutations and fusions of BRAF with KIAA1549 or FAM131B are characteristics of pilocytic astrocytomas. In 2008, a tandem duplication was confirmed in 7q34, and a new fusion gene was found to be generated by a fusion between the KIAA1549 gene and BRAF genes, which was previously uncharacterized in pilocytic astrocytomas [58]. Fusions between KIAA1549 exon 15 and BRAF exon 9 and those between FAM131B exon 2 and BRAF exon 9 are derived from a 2.0 Mb tandem duplication in chromosome band 7q34, which can be detected by realtime polymerase chain reaction (PCR) or fluorescent in situ hybridization (FISH) (Fig. 2) [62].
- The BRAF V600E mutation was found in two-thirds of all pleomorphic xanthoastrocytomas, one-fourth of gangliogliomas, and one-seventh of pilocytic astrocytomas [35].
- In children, CDKN2A deletions and BRAF mutations are early events in low-grade gliomas (pediatric low-grade glioma [PLGG]) undergoing malignant transformation (Fig. 2) [96]. The BRAF V600E mutant PLGG has longer transformation latency periods than the BRAF wild type PLGG (median latency period, 6.65 years vs 1.59 years, respectively) [96]. As a result, all of the patients with secondary high-grade glioma (sHGGs) containing mutant BRAF were diagnosed at age 9 or older [96]. The sHGG in children showed recurrent changes of BRAF V600E mutations and CDKN2A deletions in 39% and 57%, respectively.
- TERTp mutations
- Human telomerase is a ribonucleoprotein that regulates the length of telomeric DNA at the ends of chromosomes; therefore, it plays an important role in cellular immortalization and oncogenesis [13]. Mutations in TERTp are molecular hallmarks of glioma, occurring in more than 80%–96% of IDH-wildtype GBMs and oligodendrogliomas, but less frequently present in grade II and III astrocytomas (38.5%) (Table 1, Fig. 1) [97]. Chan et al. (2015) [98] found that mutations in TERTp are present in 41 of 142 (28.9%) grade II gliomas and 20 of 72 (27.8%) grade III gliomas. The mutations were always found in two hotspots (chr5: 1 295 228 C > T and 1 295 250 C > T), resulting in a somatic gain-of-function mutation and an enhancement of TERTp activity [99]. A relative telomere length is strongly correlated with TERTp mutations. These two hot spot mutations are mutually exclusive in gliomas and TP53 mutations, but coincident with a 1p/19q codeletion [100].
- The prognostic effect of the TERTp mutation is controversial. Some studies have suggested that TERTp mutations in low-grade gliomas are associated with shorter progression-free survival [101]. Other researchers have found that TERTp mutations are a predictor of a poorer response to temozolomide [102]. TERTp mutations are associated with good outcome in gliomas with mutant IDH, but it is also related to poor outcome in gliomas with wild type IDH [97]. In gliomas with wildtype IDH or GBMs with hypomethylated MGMT, TERTp mutations were found to predict poor prognosis [103]. Therefore, when using the TERTp mutational status as a prognostic factor, other factors such as mutations in IDH and tumor grade should be considered [101]. Primer sequences for TERTp mutation is summarized in Table 5.
- C11orf95-RELA fusion
- More than two-thirds of supratentorial ependymomas contain oncogenic fusions between RELA (the principle effector of canonical nuclear factor κB [NF-κB] signaling) and C11orf95 (an uncharacterized gene) (Table 2) [32,34]. Ependymomas carrying the C11orf95-RELA fusion are characterized by a nuclear accumulation of p65RelA (NFκB) indicating a pathological activation of the NFkB signaling pathway [34]. C11orf95-RELA fusions result from chromothripsis involving chromosome 11q13.1. Furthermore, the C11orf95-RELA fusion protein was found to spontaneously translocate to the nucleus to activate NF-kB target genes, and to rapidly transform neural stem cells, the cell of origin for ependymomas, to form these tumors in mice, which is a poor prognostic marker (Table 2) [34].
- CTNNB1 mutations
- Mutations in CTNNB1 on chromosome 3p21, which encodes β-catenin, promote the stabilization and nuclear translocation of itself, therefore activating the WNT signaling pathway (Table 2). The nuclear translocation of β-catenin is present in medulloblastomas, solitary fibrous tumors/hemangiopericytomas, and adamantinomatous-type craniopharyngiomas. Nuclear expression of β-catenin is always focal. Overall, these mutations can act as future therapeutic targets in nuclear β-catenin–positive tumors.
- MYC and MYCN amplification
- MYC is a regulator gene that encodes for transcription factors for c-myc, Mycn, and Mycl [77]. Nuclear myc protein has multiple functions such as cell cycle progression, apoptosis, and cellular transformation. MYC/MYCN amplification or overexpression is found not only in type-3 medulloblastomas, but also in other aggressive subtypes of medulloblastomas, which account for approximately 10% of medulloblastomas [78,104].
- SMARCB1 and SMARCA4 mutations
- AT/RT are highly malignant CNS embryonal tumors with rhabdoid morphology, where biallelic inactivation of SMARCB1 results in a loss of INI1 (BAF47) nuclear expression (Tables 2–4). The loss of BRG1 nuclear expression in AT/RT with mutations in SMARCA4 is rarely reported [105]. The tumor suppressors SMARCB1 and SMARCA4 are located on 22q11.23 and 19p13.2, respectively.
- The biallelic loss of SMARCB1 and SMARCA4 can be recognized by nuclear negativity of the INI1 and BRG1 immunostaining. However, FISH and DNA sequencing can also be used to detect these genetic alterations.
- WES and RNA sequencing of AT/RT revealed few somatic mutations and several deregulated signaling pathways related to the SMARCB1 deficiency [106].
- C19MC amplification
- Chromosomal 19 micro-ribosomal clusters (C19MC), localized at 19q13.42, are produced by chromothripsis, a phenomenon of chromosomal breakage and inaccurate recombination, resulting in the rearrangement of thousands of clustered chromosomes in a single event to a localized and restricted genomic region on one or more chromosomes [107]. C19MC amplification is a genetic feature of embryonal tumors with multilayer rosettes (ETMR), supratentorial ependymomas, and medulloepitheliomas (Table 2) [108]. This abnormality can be detected by FISH.
- MGMT promoter hypermethylation
- One of the most clinically important DNA methylation markers in GBMs is the promoter of MGMT, a DNA repair enzyme that can abrogate the effects of alkylating chemotherapy such as temozolomide. More than 50% of oligodendrogliomas and 30%–40% of adult high-grade gliomas (approximately 40% of IDH-wildtype GBMs) reveal MGMT methylation. MGMT promoter hypermethylation induces gene silencing and susceptibility to combined temozolomide and radiation therapy. In cases of oligodendroglioma with promoter methylation of the MGMT gene, whether or not PCV chemotherapy is advantageous remains controversial [59]. However, it is still considered to be the most accurate predictive factor for survival during PCV chemotherapy [60].
IMPORTANT MOLECULAR GENETICS FOUND IN BRAIN TUMORS
TP53 mutation
- The molecular testing methods commonly used in pathology laboratories include immunohistochemical staining, direct sequencing, FISH, chromosomal genomic hybridization, and NGS.
- Immunohistochemical studies
- Some of the genetic changes can be detected by immunohistochemical studies. IDH R132H, H3 K27M, BRAF V600E, and EGFRvIII can be diagnosed using mutation-specific monoclonal antibodies (mAbs). IDH1 (H09), K27M, and BRAF VE1 antibodies have 100% sensitivity and specificity to detect gene mutations [36]. The INI-1 (SMARCB1), BRG1 (SMARCA4), p16 (CDKN2A), CIC, and FUBP1 mutations and MGMT methylation can be detected by the loss of expression. TP53 mutations are detected by the complete loss of expression or overexpression via the stabilization of the mutant protein. In addition, STAT6 (NAB2-STAT6 fusion), L1CAM (protein marker for RELA-c9ORF95 fusion), and the amplifications of EGFR, MET, and PDGFR can be analyzed by overexpression assays (Table 4) [21,24,36,55]. Mutations in several genes give rise to the overexpression or loss of proteins, depending on whether the mutation is a gain of function or a loss of function mutation, respectively. For example, p53 nuclear overexpression reflects a TP53 mutation, and p16 nuclear and cytoplasmic losses are associated with a CDKN2A homozygous deletion. Furthermore, the nuclear loss of ATRX is associated with an ATRX mutation, and MLH1, MSH2, and PMS nuclear losses are associated with methylation of these genes. Among these, all but TP53 have high correlations between immunohistochemical results and molecular genetic studies. The correlation rate of p53 between immunohistochemistry and mutation studies is low. This is because p53 immunoreactivity may have resulted from the prolongation of half-life either due to p53 mutations or the accumulation of wild type protein brought about by mechanisms other than those caused by mutations, such as a complex formation with MDM2 overexpression products [109], whereas p53 negativity can be observed by the methylation of the TP53 promoter. Therefore, overexpression of p53 protein is not always associated with mutations in conserved exons of the p53 gene.
- Direct sequencing
- The rapidly growing number of prognostic and predictive genetic markers in neuro-oncology has led to an increasing need for a more thorough molecular analysis of brain tumor specimens in a modern pathology setting. Although several diagnostically important mutations can be detected by immunohistochemistry, this is not the case for the full spectrum of alterations now known to be of relevance. Therefore, the mutations of IDH 1/2, BRAF, CTNNB1, H3F3A (K27M, G34), TP53, ATRX, and TERTp are usually detected by direct sequencing. The sequencing conditions, primer sequences, and PCR product sizes that are currently being used for brain tumor diagnosis are summarized in Table 5.
- In order to detect mutations in CIC (missense mutations or small in-frame deletions) and FUBP1 (indel, splicing alteration, and nonsense mutations) in the functional regions such as the HMG box and CI motif, NGS is required because the mutation sites of these two genes are widely distributed along the coding regions [88].
- Sanger sequencing is a method of DNA sequencing based on the selective incorporation of chain-terminating dideoxynucleotides by DNA polymerase during DNA replication in vitro, which was developed by Frederick Sanger and colleagues in 1977 [91].
- Recently, one-step Sanger sequencing (combining the amplification and sequencing steps) methods such as Ampliseq and SeqSharp have been developed that allows for rapid sequencing of target genes without cloning or prior amplification.
- However, covering all potentially clinically relevant genes in a routine diagnostic setting by these methods has become virtually impossible. Reliable high-throughput methods allowing for parallel analysis of multiple targets emerged as an attractive alternative for comprehensive diagnostic neuropathology. NGS provides opportunities to evaluate many genomic targets with high accuracy and sensitivity due to high sequencing coverage. However, the Sanger method is widely used for small-scale projects, and validation of NGS results, especially long serial DNA sequence reads that are greater than 500 nucleotides.
- Pyrosequencing
- Pyrosequencing is a method of DNA sequencing based on the “sequencing by synthesis” principle [110]. It differs from Sanger sequencing in that it relies on the detection of pyrophosphates that are released upon nucleotide incorporation rather than chain termination with dideoxynucleotides. Various gene alterations can be detected by pyrosequencing, including IDH1/IDH2 mutations, TERTp mutation and MGMT methylation [92,111-113].
- NGS
- More recently, high volume Sanger sequencing has been supplanted by NGS methods, especially for large-scale, automated genome analyses. NGS is a high-capacity parallel sequencing process that handles hundreds to millions of DNA fragments simultaneously [114,115]. Currently, there are second- and third-generation sequencing technologies that continue to reduce the cost of DNA sequencing and improve accuracy. The ability to multiplex several samples also leads to increased throughput and reduced cost. Soon, NGS will enter clinical practices but it is still uncertain whether it can replace the established techniques. Depending on the type of NGS panel, it can only detect single nucleotide variations and indels or embrace CNV and translocations. The performance of the NGS panel requires rigorous validations and strict quality control for its sensitivity, specificity, and accuracy. There are several NGS panels for brain tumor companion diagnosis [78,79,95,116]. Current practice and guidelines for the clinical use of a NGS-based oncology test are described in Strom’s 2016 paper [117]. A comparison of different NGS-based diagnostic tools is summarized in Lapin et al. paper (2016) [118].
- FISH
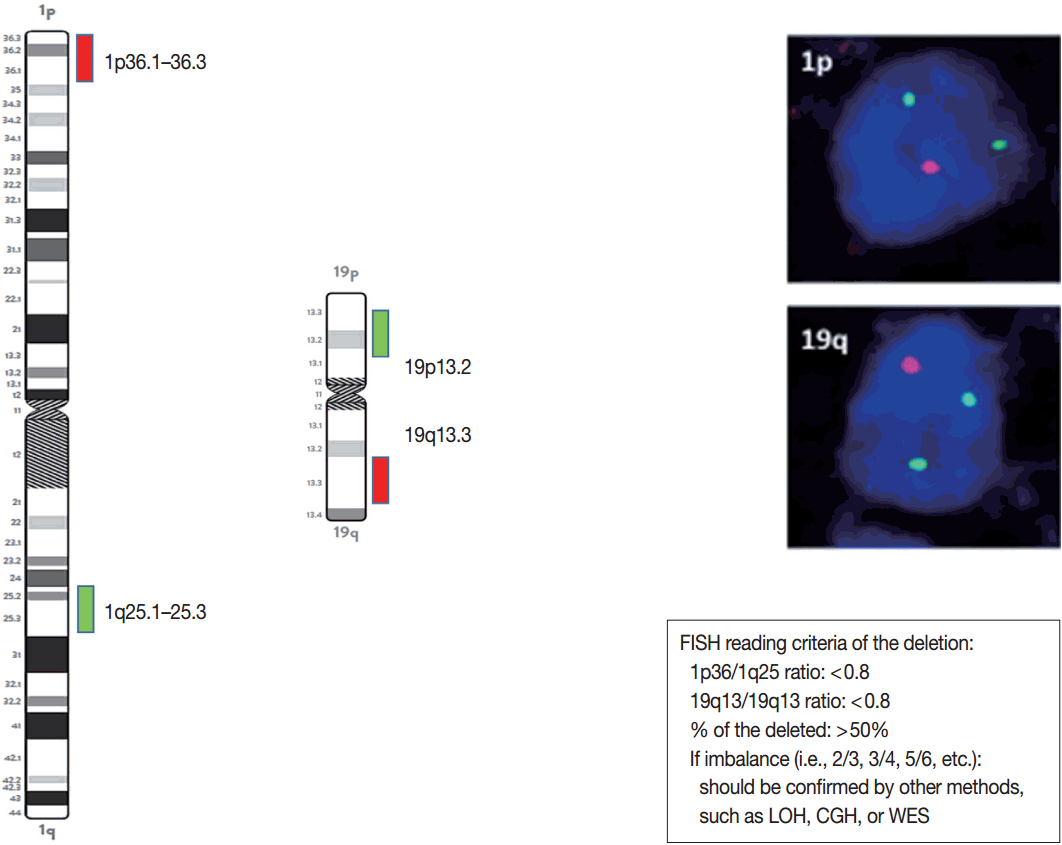
- FISH studies can be easily used to identify chromosomal aberrations, such as a 1p/19q codeletion, EGFR amplification or high polysomy, PTEN and CDKN2A homozygous or hemizygous deletions, C-MYC and N-MYC amplifications, BRAF fusions or copy gains, C11orf95-RELA fusions, C11orf95-YAP1 fusions, YAP1-MAMLD1 fusions, and MYB-QKI fusions. The probes and reading criteria of currently used FISH for the diagnosis and prognosis of brain tumors are summarized in Table 6.
- The presence or absence of the 1p/19q codeletion can be determined by FISH, comparative genomic hybridization (CGH), or microsatellite analyses for a loss of heterozygosity (LOH) on chromosomes 1p and 19q [119,120].
- When the amount of DNA is insufficient to carry out the single nucleotide polymorphism or CGH array, microsatellite analysis is performed to evaluate via PCR to determine whether there is a LOH of chromosome 1p and 19q [121]. In cases of 1p/19q codeletion, the evaluation and interpretation of FISH results are based on International Society of Pediatric Oncology (SIOP) guidelines for neuroblastoma study [80]. For each hybridization, the number of green and orange signals is assessed for a minimum of 100 nonoverlapping nuclei. An interpretation of a deletion was made when > 50% of nuclei harbored less than a single orange signal (0/2, 1/2, 0/3, 1/3, etc). Such deletions most likely correspond to a LOH (Fig. 3). However, with increasing grades of malignancy, genomic polyploidies may be encountered. These chromosomal polysomies may be balanced (e.g., 3/3, 4/4, 5/5, etc.) or imbalanced (e.g., 3/2, 4/2, 3/5, 4/5, etc.), indicating relative gains or losses. Whether an imbalance situation with relative loss of the target 1p or 19q corresponds to a hemizygous deletion in the presence of reduplication cannot be solved by FISH. In such cases, secondary assays such as PCR-based LOH or CGH studies should be used for better specificity [80].
- Methylation specific PCR (O6 methyl guanine methyl transferase)
- Methylation can be detected by methylation specific PCR (MSP), pyrosequencing, multiplex ligation-dependent probe assay, or BeadChip microarray [57,113,122]. In the Seoul National University Hospital, MSP is conducted using an EZ DNA methylation Kit (catalog No. D5005, Zymo Research, Irvine, CA, USA) to determine the methylation status of the MGMT promoter. The primer set is described in Table 5. The primer sequences for MGMT were as follows: methylated forward, 5′-TTT CGA CGT TCG TAG GTT TTC GC-3′; methylated reverse, 5′-GCA CTC TTC CGA AAA CGA AACG 3′; unmethylated forward, 5′-TTT GTG TTT TGA TGT TTG TAG GTT TTT GT-3′; and unmethylated reverse, 5′-AAC TCC ACA CTC TTC CAA AAA CAA AAC A-3′.
METHODS OF MOLECULAR TESTING
- Among primary adult and pediatric CNS tumors, diffuse gliomas are the largest and most diverse group. They usually arise in the cerebral hemispheres and are defined by their widely infiltrative properties and tendency for biological progression. According to the 2016 WHO criteria, gliomas can be classified using combined histological and molecular markers as diffuse astrocytic and oligodendroglial tumors, other astrocytic tumors, ependymal tumors, or other gliomas. The most aggressive astrocytic tumors with a dismal prognosis are the GBMs with wild type IDH and DMG, histone-mutant (H3 K27, G34, and K36). Less infiltrative gliomas in children and young adults include WHO grade I pilocytic astrocytomas,WHO grade II/III pleomorphic xanthoastrocytomas, and WHO grade I subependymal giant cell astrocytomas. Other major classes of CNS tumors include neuronal and mixed glioneuronal tumors as well as embryonal tumors such as cerebellar medulloblastomas, and AT/RT, and extra-axial meningiomas.
- The diagnosis of CNS tumors is historically primarily based on histopathological features. However, studies have shown that patients with morphologically identical tumors have different clinical outcomes and treatment responses due to the different molecular genetic characteristics of the tumor. Therefore, many molecular markers became deeply integrated into the diagnosis of CNS tumors and are now used to guide prognosis and treatment of patients. If molecular genetic testing is not possible, a diagnosis is made with the nonspecific term “glioma, not otherwise specified (NOS).” In this case, because the grade and the exact diagnosis of the tumor as well as the biological markers are covered, the precision diagnosis and the appropriate treatments are no longer possible. These “NOS diagnostics” must be reclassified using molecular genetic diagnostic tools.
- Diffuse astrocytic and oligodendroglial tumors
- Molecular genetic testing can classify diffuse astrocytic and oligodendroglial tumors (WHO grades II–III) and differentiate IDH-mutant GBMs from IDH-wildtype GBMs based on molecular signature of astrocytic and oligodendrogial tumors. Diffuse astrocytic tumors and IDH-mutant GBMs are characterized by mutations in IDH1/2, TP53, and ATRX, whereas oligodendrogliomas are characterized by mutations in IDH1/2, CIC, FUBP1, and TERTp, and a codeletion in 1p/19q (whole chromosomal arm deletion) (Fig. 1). IDH-wildtype GBMs can be identified by demonstrating a dysregulation of several critical signaling pathways and a lack of the above-mentioned genetic alterations except TERTp mutations, which are common in IDH-wildtype GBM. The main signaling pathways involved in IDH-wildtype GBMs are RTK/RAS/PI3K pathways (via amplification and mutations in EGFR [EGFRvIII], PIK3CA, RAS, NF1, and MET) and TP53 and RB1 suppressor pathways (via mutations in or loss of TP53, CDKN2A, and RB1 genes) (Fig. 1) [94].
- Mutations in IDH1/2 should be studied in diffuse gliomas for the WHO classification of CNS tumors. They are favorable prognostic markers and are genomic abnormalities initially present in both astrocytic and oligodendroglial tumors. Therefore, from the revised version of 2016 fourth edition of the CNS Tumor Classification, these two tumor categories have been combined as diffuse astrocytic and oligodendroglial tumors [7]. P53 and ATRX mutations are now the hallmark of WHO grade II and grade III astrocytic tumors. TERTp mutations, such as C228T and C250T, are found in more than 90% of oligodendroglial tumors and in more than 80% of GBMs as well as about 30%–40% of lower grade gliomas. Mutations in TERTp were associated with poor survival and resistance to radiotherapy in GBMs and in lower grade gliomas (grade II and III) without IDH mutation; however, TERTp mutations were associated with a favorable prognosis in lower grade gliomas with IDH mutations [63,123].
- High- and low-grade pediatric gliomas
- Pediatric high-grade gliomas are unique, featuring mutations in H3F3A, ATRX, and DAXX, whereas PLGGs contain mutations or rearrangements in BRAF, FGFR1, or MYB. Furthermore, mutations in IDH are rare unless the patient is an adolescent (Fig. 2) [22,64,65]. The circumscribed low-grade gliomas such as pilocytic astrocytoma, pilomyxoid astrocytoma, ganglioglioma, and PXA often harbor the BRAF V600E or FGFR1 mutation, or gene fusions involving BRAF with KIAA1549 or FAM131B [35,62,65]. Histone H3 K27M, K36, and G34 mutations should be studied in pediatric glioma arising in the midline of CNS, i.e., the thalamus, pons, and spinal cord [124]. However, the histone G34 mutation can be present in cerebral high-grade diffuse gliomas of the adult (about 5%) and the histone K36 mutation is rarely found in pediatric midline gliomas [84].
- Finally, the BRAF V600E mutation can be studied in low-grade glioneuronal tumors, such as PXA (about 60%), ganglioglioma (about 25%), pilocytic astrocytoma (about 15%), epithelioid GBM (about 50%), and papillary type of craniopharingioma (about 100%) [35,66].
- Medulloblastomas and other embryonic tumors
- Medulloblastomas have been recently divided into several subtypes based on specific driver mutations including WNT, SHH, group 3, and group 4 [125-127]. Mutations in CTNNB1, DDX3X or monosomy 6 in the Wnt pathway of medulloblastomas tend to lead to a much better prognosis. In contrast, MYC or MYCN/CDK6 amplification is characteristic of group 3 and 4 medulloblastomas, which are far more likely to metastasize and have a poor prognosis even with intensive therapy. Tumors with PTCH and SMO belong to the SHH class, and have a relatively intermediate prognosis between Wnt and group 3/4 tumors. CTNNB1 mutations can be present in classical-type medulloblastomas and adamantinomatous-type craniopharyngiomas [61,125,128].
- SMARCB1 (INI1) or SMARCA4 (BRG1) gene mutations or deletions are essential for the diagnosis of AT/RT, which show dismal prognosis [44-46]. The molecular genetic hallmark of ETMR is chromosome 19 miRNA cluster (C19MC) amplification [129]. If certain tumors are morphologically similar to these tumors, i.e., rhabdoid feature, but molecular studies do not reveal these genetic abnormalities or molecular studies cannot be done, CNS embryonal tumor with rhabdoid feature or ETMR, NOS can be made as a pathological diagnosis [7,129]. Therefore, the molecular characterization of medulloblastomas and embryonal tumors have lasting clinical value.
- Meningioma and meningeal solitary fibrous tumor/hemangiopericytoma
- Meningiomas often have mutations in the NF2, AKT1, SMO, and KLF4 genes. Furthermore, meningiomas with mutant NF2 are far more likely to exhibit atypical grade II features than the other subtypes [50-52]. Recurrent mutations in KLF4, AKT1, and SMO genes are often present in NF2-negative sporadic meningiomas [50]. Clinical trials involving SMO/AKT/NF2 inhibitors are open for patients with progressive meningiomas with SMO/AKT/NF2 mutations [50,81].
- Meningeal hemangiopericytomas and solitary fibrous tumors have the same genetic modification as NAB2-STAT6 gene fusion and are considered as the same tumor [48]. This gene fusion study is essential for the differential diagnosis of histologically similar tumors.
- Genetic alterations as prognostic and predictive markers and therapeutic targets
- Overall, the analysis of multiple molecular markers not only aids in establishing a correct morphological diagnosis, but also highlights the biological differences between morphologically similar tumors. It can also help with the clinical management of patients. For example, patients with IDH1/TERTp/CIC/FUBP1 positive lower grade gliomas (WHO grades II–III) have a significantly longer median overall survival than those with IDH1/TP53/ATRX mutations [86,130].
- In addition, several molecular biomarkers showed promise in predicting responses to targeted therapies [94,131]. For example, clinical trials are now open for GBMs with EGFRvIII mutations, vemurafenib is being evaluated in BRAF V600E mutant gliomas, and clinical response has already been observed in FGFR3-TACC3–positive patients treated with an FGFR inhibitor [78]. NGS-based high-throughput molecular testing may provide potential new targets for precision medicine.
DISCUSSION
- We are now in the era of precision medicine, and have slowly caught up with the rapid development of precision medicine. High-throughput assays that can test multiple genes at once are essential for pathological diagnosis in daily practice, and NGS technology has made this possible. Current practices and guidelines for the clinical use of NGS-based oncology tests are well documented in Strom’s paper [117]. However, the medical insurance system has not kept up with the speed of technical development, the advances in diagnostic modalities, and newly developed treatment options. These are obstacles that prevents the doctors from implementing the precision medicine. In order to introduce the advanced knowledge and technologies into the daily practice of pathological diagnosis and clinical fields and reduce the costs, the medical insurance system should be formulated accordingly.
CONCLUSION
-
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Notes
Acknowledgments
LGG, low grade glioma; PXA, pleomorphic xanthoastrocytoma; GG, ganglioglioma; DA, diffuse astrocytoma; PA, pilocytic astrocytoma; AG, angiocentric glioma; DNT, dysembryoplastic neuroepithelial tumor; HGG, high grade glioma; ST, supratentorial; PF, posterior fossa; E, ependymoma; WNT, wingless signaling pathway; i, isochromosome; AT/RT, atypical teratoid/rhabdoid tumor; ETMR, embryonal tumor with multilayer rosettes.
PA, pilocytic astrocytoma; PXA, pleomorphic xanthoastrocytoma; DA, diffuse astrocytoma; AA, anaplastic astrocytoma; GBM, glioblastoma; DMG, diffuse midline glioma; HGG, high grade glioma; ODG, oligodendroglioma; MB, medulloblastomas; AT/RT, atypical teratoid/ rhabdoid tumor; WHO, World Health Organization; IHC, immunohistochemistry; FISH, fluorescent in situ hybridization; CGH, comparative genomic hybridization; RT-PCR, reverse transcription polymerase chain reaction; -, not known; MSP-PCR, methylation-specific polymerase chain reaction.
| Antibody | Positive loci | Mutated gene status | Positive result | Tumors |
|---|---|---|---|---|
| ATRX | Nucleus | Loss of function mutation | Negative | Diffuse and anaplastic astrocytoma |
| β-Catenin | Nucleus | Gain of function mutation | Focal positive | WNT type medulloblastoma, adamantinomatous craniopharyngioma |
| BRAF VE1 (BRAF V600E) | Cytoplasm | Gain of function mutation | Positive | Pleomorphic xanthoastrocytoma, ganglioglioma, pilocytic astrocytoma, epithelioid glioblastoma, papillary craniopharyngioma |
| BRG1 | Nucleus | Gain of function mutation | Negative | Atypical teratoid rhabdoid tumor |
| CIC | Nucleus | Loss of function mutation | Negative | Oligodendroglioma (47%) [15] |
| c-MET | Membrane | Overexpression | Positive | Glioblastoma, anaplastic astrocytoma |
| EGFR | Membrane | Overexpression | Positive | Glioblastoma, anaplastic astrocytoma |
| EGFRvIII | Membrane | Overexpression | Positive | Glioblastoma, anaplastic astrocytoma |
| H3 K27M | Nucleus | Gain of function mutation | Positive | Diffuse midline glioma |
| FUBP1 | Nucleus | Loss of function mutation | Negative | Oligodendroglioma (16%) [15] |
| IDH1 (H09) | Nucleus and cytoplasm | Gain of function mutation | Positive | Astrocytoma and oligodendroglioma |
| INI1 | Nucleus | Loss of function mutation | Negative | Atypical teratoid rhabdoid tumor |
| P16 | Nucleus and cytoplasm | Loss of function mutation | Negative | High grade glioma |
| P53 | Nucleus | Overexpression | Positive | Astrocytic tumors |
| PDGFRA | Membrane | Overexpression | Positive | Glioblastoma, anaplastic astrocytoma |
| PTEN | Cytoplasm | Loss of function mutation | Negative | Glioblastoma |
| RELA (NFKB3) | Cytoplasm | Gain of function mutation | Positive | Cerebral ependymoma |
| STAT6 | Nucleus | Gain of function mutation | Positive | Solitary fibrous tumor/hemangiopericytoma |
| MLH1 | Nucleus | Loss of function mutation | Negative | Gliomas |
| MSH2 | Nucleus | Loss of function mutation | Negative | Gliomas |
| PMS2 | Nucleus | Loss of function mutation | Negative | Gliomas |
| Ki67 | Nucleus | Overexpression | Positive | Brain tumors (for an ancillary test for tumor grading) |
| pHH3 | Nucleus | Overexpression | Positive | Brain tumors (for counting mitoses) |
| GFAP | Nucleus | Expression | Positive | Astrocytic tumors |
| Olig2 | Nucleus | Expression | Positive | Gliomas including astrocytomas and oligodendroglioma |
| Neither neuronal nor ependymal tumors |
- 1. Lee CH, Jung KW, Yoo H, Park S, Lee SH. Epidemiology of primary brain and central nervous system tumors in Korea. J Korean Neurosurg Soc 2010; 48: 145–52. ArticlePubMedPMC
- 2. Jung KW, Ha J, Lee SH, Won YJ, Yoo H. An updated nationwide epidemiology of primary brain tumors in republic of Korea. Brain Tumor Res Treat 2013; 1: 16–23. ArticlePubMedPMC
- 3. Cancer Genome Atlas Research Network, Brat DJ, Verhaak RG, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 2015; 372: 2481–98. ArticlePubMedPMC
- 4. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell 2013; 155: 462–77. ArticlePubMedPMC
- 5. Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006; 9: 157–73. ArticlePubMed
- 6. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008; 321: 1807–12. ArticlePubMedPMC
- 7. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 2016; 131: 803–20. ArticlePubMedPDF
- 8. Weller M, Weber RG, Willscher E, et al. Molecular classification of diffuse cerebral WHO grade II/III gliomas using genome- and transcriptome-wide profiling improves stratification of prognostically distinct patient groups. Acta Neuropathol 2015; 129: 679–93. ArticlePubMed
- 9. Rajmohan KS, Sugur HS, Shwetha SD, et al. Prognostic significance of histomolecular subgroups of adult anaplastic (WHO Grade III) gliomas: applying the ‘integrated’ diagnosis approach. J Clin Pathol 2016; 69: 686–94. ArticlePubMed
- 10. Malzkorn B, Reifenberger G. Practical implications of integrated glioma classification according to the World Health Organization classification of tumors of the central nervous system 2016. Curr Opin Oncol 2016; 28: 494–501. ArticlePubMed
- 11. Clark K, Voronovich Z, Horbinski C. How molecular testing can help (and hurt) in the workup of gliomas. Am J Clin Pathol 2013; 139: 275–88. ArticlePubMedPMC
- 12. Reuss DE, Sahm F, Schrimpf D, et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol 2015; 129: 133–46. ArticlePubMed
- 13. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol 2015; 129: 829–48. ArticlePubMed
- 14. Bush NA, Chang SM, Berger MS. Current and future strategies for treatment of glioma. Neurosurg Rev 2017; 40: 1–14. ArticlePubMedPDF
- 15. Chan AK, Pang JC, Chung NY, et al. Loss of CIC and FUBP1 expressions are potential markers of shorter time to recurrence in oligodendroglial tumors. Mod Pathol 2014; 27: 332–42. ArticlePubMed
- 16. Kreth S, Thon N, Kreth FW. Epigenetics in human gliomas. Cancer Lett 2014; 342: 185–92. ArticlePubMed
- 17. Ostrom QT, Gittleman H, Stetson L, Virk SM, Barnholtz-Sloan JS. Epidemiology of gliomas. Cancer Treat Res 2015; 163: 1–14. ArticlePubMed
- 18. Reis GF, Pekmezci M, Hansen HM, et al. CDKN2A loss is associated with shortened overall survival in lower-grade (World Health Organization Grades II-III) astrocytomas. J Neuropathol Exp Neurol 2015; 74: 442–52. ArticlePubMedPMC
- 19. Simeonova I, Huillard E. In vivo models of brain tumors: roles of genetically engineered mouse models in understanding tumor biology and use in preclinical studies. Cell Mol Life Sci 2014; 71: 4007–26. ArticlePubMedPMC
- 20. Cachia D, Kamiya-Matsuoka C, Mandel JJ, et al. Primary and secondary gliosarcomas: clinical, molecular and survival characteristics. J Neurooncol 2015; 125: 401–10. ArticlePubMedPMC
- 21. Louis DN, Perry A, Burger P, et al. International Society of Neuropathology: Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 2014; 24: 429–35. ArticlePubMedPMC
- 22. Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482: 226–31. ArticlePubMed
- 23. Khuong-Quang DA, Buczkowicz P, Rakopoulos P, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 2012; 124: 439–47. ArticlePubMedPMC
- 24. Solomon DA, Wood MD, Tihan T, et al. Diffuse midline gliomas with histone H3-K27M mutation: a series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol 2016; 26: 569–80. ArticlePubMed
- 25. Zhang R, Han J, Daniels D, Huang H, Zhang Z. Detecting the H3F3A mutant allele found in high-grade pediatric glioma by real-time PCR. J Neurooncol 2016; 126: 27–36. ArticlePubMed
- 26. Nambirajan A, Malgulwar PB, Sharma MC, et al. C11orf95-RELA fusion present in a primary intracranial extra-axial ependymoma: report of a case with literature review. Neuropathology 2016; 36: 490–5. ArticlePubMed
- 27. Wu J, Armstrong TS, Gilbert MR. Biology and management of ependymomas. Neuro Oncol 2016; 18: 902–13. ArticlePubMedPMC
- 28. Figarella-Branger D, Lechapt-Zalcman E, Tabouret E, et al. Supratentorial clear cell ependymomas with branching capillaries demonstrate characteristic clinicopathological features and pathological activation of nuclear factor-kappaB signaling. Neuro Oncol 2016; 18: 919–27. ArticlePubMedPMC
- 29. Nobusawa S, Hirato J, Sugai T, et al. Atypical teratoid/rhabdoid tumor (AT/RT) arising from ependymoma: a type of AT/RT secondarily developing from other primary central nervous system tumors. J Neuropathol Exp Neurol 2016; 75: 167–74. ArticlePubMed
- 30. Olar A, Sulman EP. Molecular markers in low-grade glioma-toward tumor reclassification. Semin Radiat Oncol 2015; 25: 155–63. ArticlePubMedPMC
- 31. Capper D, Reifenberger G. Classification of gliomas: current progress and perspectives. Nervenarzt 2015; 86: 672–83. ArticlePubMed
- 32. Cachia D, Wani K, Penas-Prado M, et al. C11orf95-RELA fusion present in a primary supratentorial ependymoma and recurrent sarcoma. Brain Tumor Pathol 2015; 32: 105–11. ArticlePubMed
- 33. Nobusawa S, Hirato J, Yokoo H. Molecular genetics of ependymomas and pediatric diffuse gliomas: a short review. Brain Tumor Pathol 2014; 31: 229–33. ArticlePubMed
- 34. Parker M, Mohankumar KM, Punchihewa C, et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 2014; 506: 451–5. ArticlePubMedPMC
- 35. Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 2011; 121: 397–405. ArticlePubMed
- 36. Tanboon J, Williams EA, Louis DN. The diagnostic use of immunohistochemical surrogates for signature molecular genetic alterations in gliomas. J Neuropathol Exp Neurol 2016; 75: 4–18. ArticlePubMed
- 37. Thomas L, Di Stefano AL, Ducray F. Predictive biomarkers in adult gliomas: the present and the future. Curr Opin Oncol 2013; 25: 689–94. ArticlePubMed
- 38. Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013; 45: 602–12. ArticlePubMedPMC
- 39. Korshunov A, Meyer J, Capper D, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 2009; 118: 401–5. ArticlePubMed
- 40. Tian Y, Rich BE, Vena N, et al. Detection of KIAA1549-BRAF fusion transcripts in formalin-fixed paraffin-embedded pediatric low-grade gliomas. J Mol Diagn 2011; 13: 669–77. ArticlePubMedPMC
- 41. Cin H, Meyer C, Herr R, et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol 2011; 121: 763–74. ArticlePubMed
- 42. Collins VP, Jones DT, Giannini C. Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol 2015; 129: 775–88. ArticlePubMedPMC
- 43. Roth JJ, Fierst TM, Waanders AJ, Yimei L, Biegel JA, Santi M. Whole chromosome 7 gain predicts higher risk of recurrence in pediatric pilocytic astrocytomas independently from KIAA1549-BRAF fusion status. J Neuropathol Exp Neurol 2016; 75: 306–15. ArticlePubMedPMC
- 44. Sredni ST, Tomita T. Rhabdoid tumor predisposition syndrome. Pediatr Dev Pathol 2015; 18: 49–58. ArticlePubMed
- 45. Hasselblatt M, Gesk S, Oyen F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 2011; 35: 933–5. ArticlePubMed
- 46. Fruhwald MC, Biegel JA, Bourdeaut F, Roberts CW, Chi SN. Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol 2016; 18: 764–78. ArticlePubMedPMC
- 47. Diamandis P, Ferrer-Luna R, Huang RY, et al. Case report: next generation sequencing identifies a NAB2-STAT6 fusion in glioblastoma. Diagn Pathol 2016; 11: 13.ArticlePubMedPMCPDF
- 48. Han N, Kim H, Min SK, et al. Meningeal solitary fibrous tumors with delayed extracranial metastasis. J Pathol Transl Med 2016; 50: 113–21. ArticlePubMedPDF
- 49. Maekawa A, Kohashi K, Yamada Y, et al. A case of intracranial solitary fibrous tumor/hemangiopericytoma with dedifferentiated component. Neuropathology 2015; 35: 260–5. ArticlePubMed
- 50. Brastianos PK, Horowitz PM, Santagata S, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet 2013; 45: 285–9. ArticlePubMedPMC
- 51. Clark VE, Erson-Omay EZ, Serin A, et al. Genomic analysis of non NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013; 339: 1077–80. ArticlePubMedPMC
- 52. Reuss DE, Piro RM, Jones DT, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol 2013; 125: 351–8. ArticlePubMed
- 53. Yuzawa S, Nishihara H, Tanaka S. Genetic landscape of meningioma. Brain Tumor Pathol 2016; 33: 237–47. ArticlePubMedPDF
- 54. Mehes G, Irsai G, Bedekovics J, et al. Activating BRAF V600E mutation in aggressive pediatric Langerhans cell histiocytosis: demonstration by allele-specific PCR/direct sequencing and immunohistochemistry. Am J Surg Pathol 2014; 38: 1644–8. ArticlePubMed
- 55. Rodriguez FJ, Vizcaino MA, Lin MT. Recent advances on the molecular pathology of glial neoplasms in children and adults. J Mol Diagn 2016; 18: 620–34. ArticlePubMedPMC
- 56. Maire CL, Ligon KL. Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro Oncol 2014; 16 Suppl 8: viii1–6. ArticlePubMed
- 57. Masui K, Mischel PS, Reifenberger G. Molecular classification of gliomas. Handb Clin Neurol 2016; 134: 97–120. ArticlePubMed
- 58. Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 2008; 68: 8673–7. ArticlePubMedPMC
- 59. van den Bent MJ, Dubbink HJ, Sanson M, et al. MGMT promoter methylation is prognostic but not predictive for outcome to adjuvant PCV chemotherapy in anaplastic oligodendroglial tumors: a report from EORTC Brain Tumor Group Study 26951. J Clin Oncol 2009; 27: 5881–6. ArticlePubMedPMC
- 60. Dubbink HJ, Atmodimedjo PN, Kros JM, et al. Molecular classification of anaplastic oligodendroglioma using next-generation sequencing: a report of the prospective randomized EORTC Brain Tumor Group 26951 phase III trial. Neuro Oncol 2016; 18: 388–400. ArticlePubMed
- 61. Ramaswamy V, Remke M, Bouffet E, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol 2016; 131: 821–31. ArticlePubMedPMCPDF
- 62. Roth JJ, Santi M, Pollock AN, et al. Chromosome band 7q34 deletions resulting in KIAA1549-BRAF and FAM131B-BRAF fusions in pediatric low-grade gliomas. Brain Pathol 2015; 25: 182–92. ArticlePubMed
- 63. Zhang ZY, Chan AK, Ding XJ, et al. TERT promoter mutations contribute to IDH mutations in predicting differential responses to adjuvant therapies in WHO grade II and III diffuse gliomas. Oncotarget 2015; 6: 24871–83. ArticlePubMedPMC
- 64. Bandopadhayay P, Ramkissoon LA, Jain P, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 2016; 48: 273–82. ArticlePubMedPMC
- 65. Qaddoumi I, Orisme W, Wen J, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 2016; 131: 833–45. ArticlePubMedPMCPDF
- 66. Behling F, Barrantes-Freer A, Skardelly M, et al. Frequency of BRAF V600E mutations in 969 central nervous system neoplasms. Diagn Pathol 2016; 11: 55.ArticlePubMedPMC
- 67. Appin CL, Brat DJ. Molecular pathways in gliomagenesis and their relevance to neuropathologic diagnosis. Adv Anat Pathol 2015; 22: 50–8. ArticlePubMed
- 68. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010; 465: 966.ArticlePubMedPMC
- 69. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462: 739–44. ArticlePubMedPMC
- 70. Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res 2012; 18: 5562–71. ArticlePubMedPMC
- 71. Dimitrov L, Hong CS, Yang C, Zhuang Z, Heiss JD. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int J Med Sci 2015; 12: 201–13. ArticlePubMedPMC
- 72. Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010; 17: 510–22. ArticlePubMedPMC
- 73. Appin CL, Brat DJ. Biomarker-driven diagnosis of diffuse gliomas. Mol Aspects Med 2015; 45: 87–96. ArticlePubMed
- 74. Claus EB, Walsh KM, Wiencke JK, et al. Survival and low-grade glioma: the emergence of genetic information. Neurosurg Focus 2015; 38: E6.ArticlePMC
- 75. Cohen AL, Colman H. Glioma biology and molecular markers. Cancer Treat Res 2015; 163: 15–30. ArticlePubMed
- 76. Jha P, Pia Patric IR, Shukla S, et al. Genome-wide methylation profiling identifies an essential role of reactive oxygen species in pediatric glioblastoma multiforme and validates a methylome specific for H3 histone family 3A with absence of G-CIMP/isocitrate dehydrogenase 1 mutation. Neuro Oncol 2014; 16: 1607–17. ArticlePubMedPMC
- 77. Swartling FJ. Myc proteins in brain tumor development and maintenance. Ups J Med Sci 2012; 117: 122–31. ArticlePubMedPMC
- 78. Northcott PA, Pfister SM, Jones DT. Next-generation (epi)genetic drivers of childhood brain tumours and the outlook for targeted therapies. Lancet Oncol 2015; 16: e293–302. ArticlePubMed
- 79. Blumenthal DT, Dvir A, Lossos A, et al. Clinical utility and treatment outcome of comprehensive genomic profiling in high grade glioma patients. J Neurooncol 2016; 130: 211–9. ArticlePubMedPDF
- 80. Ambros PF, Ambros IM; SIOP Europe Neuroblastoma Pathology, Biology, and Bone Marrow Group. Pathology and biology guidelines for resectable and unresectable neuroblastic tumors and bone marrow examination guidelines. Med Pediatr Oncol 2001; 37: 492–504. ArticlePubMed
- 81. Abedalthagafi M, Bi WL, Aizer AA, et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro Oncol 2016; 18: 649–55. ArticlePubMedPMC
- 82. Clynes D, Higgs DR, Gibbons RJ. The chromatin remodeller ATRX: a repeat offender in human disease. Trends Biochem Sci 2013; 38: 461–6. ArticlePubMed
- 83. Gibbons RJ, McDowell TL, Raman S, et al. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet 2000; 24: 368–71. ArticlePubMed
- 84. Chan KM, Fang D, Gan H, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013; 27: 985–90. ArticlePubMedPMC
- 85. Chen P, Zhao J, Wang Y, et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev 2013; 27: 2109–24. ArticlePubMedPMC
- 86. Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012; 3: 709–22. ArticlePubMedPMC
- 87. Bettegowda C, Agrawal N, Jiao Y, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011; 333: 1453–5. ArticlePubMedPMC
- 88. Sahm F, Koelsche C, Meyer J, et al. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol 2012; 123: 853–60. ArticlePubMed
- 89. Jiménez G, Shvartsman SY, Paroush Z. The Capicua repressor: a general sensor of RTK signaling in development and disease. J Cell Sci 2012; 125(Pt 6):1383–91. ArticlePubMedPMC
- 90. Chittaranjan S, Chan S, Yang C, et al. Mutations in CIC and IDH1 cooperatively regulate 2-hydroxyglutarate levels and cell clonogenicity. Oncotarget 2014; 5: 7960–79. ArticlePubMedPMC
- 91. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 1977; 74: 5463–7. ArticlePubMedPMC
- 92. Switzeny OJ, Christmann M, Renovanz M, Giese A, Sommer C, Kaina B. MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high-grade glioma response. Clin Epigenetics 2016; 8: 49.ArticlePubMedPMC
- 93. Tritz R, Habita C, Robbins JM, Gomez GG, Kruse CA. Catalytic nucleic acid enzymes for the study and development of therapies in the central nervous system: review article. Gene Ther Mol Biol 2005; 9A: 89–106. PubMedPMC
- 94. Venkatesan S, Lamfers ML, Dirven CM, Leenstra S. Genetic biomarkers of drug response for small-molecule therapeutics targeting the RTK/Ras/PI3K, p53 or Rb pathway in glioblastoma. CNS Oncol 2016; 5: 77–90. ArticlePubMedPMC
- 95. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol 2016; 18: 379–87. ArticlePubMed
- 96. Mistry M, Zhukova N, Merico D, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol 2015; 33: 1015–22. ArticlePubMedPMC
- 97. Yang P, Cai J, Yan W, et al. Classification based on mutations of TERT promoter and IDH characterizes subtypes in grade II/III gliomas. Neuro Oncol 2016; 18: 1099–108. ArticlePubMedPMC
- 98. Chan AK, Yao Y, Zhang Z, et al. Combination genetic signature stratifies lower-grade gliomas better than histological grade. Oncotarget 2015; 6: 20885–901. ArticlePubMedPMC
- 99. Arita H, Narita Y, Takami H, et al. TERT promoter mutations rather than methylation are the main mechanism for TERT upregulation in adult gliomas. Acta Neuropathol 2013; 126: 939–41. ArticlePubMed
- 100. Arita H, Narita Y, Fukushima S, et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol 2013; 126: 267–76. ArticlePubMed
- 101. Heidenreich B, Rachakonda PS, Hosen I, et al. TERT promoter mutations and telomere length in adult malignant gliomas and recurrences. Oncotarget 2015; 6: 10617–33. ArticlePubMedPMC
- 102. Chen C, Han S, Meng L, Li Z, Zhang X, Wu A. TERT promoter mutations lead to high transcriptional activity under hypoxia and temozolomide treatment and predict poor prognosis in gliomas. PLoS One 2014; 9: e100297. Article
- 103. Arita H, Yamasaki K, Matsushita Y, et al. A combination of TERT promoter mutation and MGMT methylation status predicts clinically relevant subgroups of newly diagnosed glioblastomas. Acta Neuropathol Commun 2016; 4: 79.ArticlePubMedPMC
- 104. Morrissy AS, Garzia L, Shih DJ, et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016; 529: 351–7. ArticlePubMedPMC
- 105. Masliah-Planchon J, Machet MC, Fréneaux P, et al. SMARCA4-mutated atypical teratoid/rhabdoid tumor with retained BRG1 expression. Pediatr Blood Cancer 2016; 63: 568–9. ArticlePubMed
- 106. Sandgren J, Holm S, Marino AM, et al. Whole exome- and mRNA sequencing of an AT/RT case reveals few somatic mutations and several deregulated signalling pathways in the context of SMARCB1 deficiency. Biomed Res Int 2015; 2015: 862039.ArticlePubMedPMCPDF
- 107. Rode A, Maass KK, Willmund KV, Lichter P, Ernst A. Chromothripsis in cancer cells: an update. Int J Cancer 2016; 138: 2322–33. ArticlePubMed
- 108. Gajjar A, Pfister SM, Taylor MD, Gilbertson RJ. Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin Cancer Res 2014; 20: 5630–40. ArticlePubMedPMC
- 109. Das A, Tan WL, Teo J, Smith DR. Glioblastoma multiforme in an Asian population: evidence for a distinct genetic pathway. J Neurooncol 2002; 60: 117–25. ArticlePubMed
- 110. Zascavage RR, Shewale SJ, Planz JV. Deep-sequencing technologies and potential applications in forensic DNA testing. Forensic Sci Rev 2013; 25: 79–105. PubMed
- 111. Cykowski MD, Allen RA, Fung KM, Harmon MA, Dunn ST. Pyrosequencing of IDH1 and IDH2 mutations in brain tumors and non-neoplastic conditions. Diagn Mol Pathol 2012; 21: 214–20. ArticlePubMed
- 112. Quillien V, Lavenu A, Ducray F, et al. Validation of the high-performance of pyrosequencing for clinical MGMT testing on a cohort of glioblastoma patients from a prospective dedicated multicentric trial. Oncotarget 2016; 7: 61916–29. ArticlePubMedPMC
- 113. Havik AB, Brandal P, Honne H, et al. MGMT promoter methylation in gliomas-assessment by pyrosequencing and quantitative methylation-specific PCR. J Transl Med 2012; 10: 36.ArticlePubMedPMC
- 114. Worst BC, van Tilburg CM, Balasubramanian GP, et al. Next-generation personalised medicine for high-risk paediatric cancer patients: The INFORM pilot study. Eur J Cancer 2016; 65: 91–101. ArticlePubMed
- 115. Sahm F, Schrimpf D, Jones DT, et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol 2016; 131: 903–10. ArticlePubMedPDF
- 116. Zacher A, Kaulich K, Stepanow S, et al. Molecular diagnostics of gliomas using next generation sequencing of a glioma-tailored gene panel. Brain Pathol 2017; 27: 146–59. ArticlePubMed
- 117. Strom SP. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol Med 2016; 13: 3–11. ArticlePubMedPMC
- 118. Lapin V, Mighion LC, da Silva CP, Cuperus Y, Bean LJ, Hegde MR. Regulating whole exome sequencing as a diagnostic test. Hum Genet 2016; 135: 655–73. ArticlePubMedPDF
- 119. Woehrer A, Sander P, Haberler C, et al. FISH-based detection of 1p 19q codeletion in oligodendroglial tumors: procedures and protocols for neuropathological practice: a publication under the auspices of the Research Committee of the European Confederation of Neuropathological Societies (Euro-CNS). Clin Neuropathol 2011; 30: 47–55. ArticlePubMed
- 120. Jha P, Sarkar C, Pathak P, et al. Detection of allelic status of 1p and 19q by microsatellite-based PCR versus FISH: limitations and advantages in application to patient management. Diagn Mol Pathol 2011; 20: 40–7. ArticlePubMed
- 121. Idbaih A, Ducray F, Dehais C, et al. SNP array analysis reveals novel genomic abnormalities including copy neutral loss of heterozygosity in anaplastic oligodendrogliomas. PLoS One 2012; 7: e45950. Article
- 122. Wiestler B, Capper D, Hovestadt V, et al. Assessing CpG island methylator phenotype, 1p/19q codeletion, and MGMT promoter methylation from epigenome-wide data in the biomarker cohort of the NOA-04 trial. Neuro Oncol 2014; 16: 1630–8. ArticlePubMedPMC
- 123. Gao K, Li G, Qu Y, et al. TERT promoter mutations and long telomere length predict poor survival and radiotherapy resistance in gliomas. Oncotarget 2016; 7: 8712–25. ArticlePubMed
- 124. Gessi M, Gielen GH, Dreschmann V, Waha A, Pietsch T. High frequency of H3F3A (K27M) mutations characterizes pediatric and adult high-grade gliomas of the spinal cord. Acta Neuropathol 2015; 130: 435–7. ArticlePubMed
- 125. Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 2011; 121: 381–96. ArticlePubMedPMC
- 126. Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 2012; 123: 473–84. ArticlePubMedPMC
- 127. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012; 123: 465–72. ArticlePubMed
- 128. Sekine S, Shibata T, Kokubu A, et al. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol 2002; 161: 1997–2001. ArticlePubMedPMC
- 129. Jakobiec FA, Kool M, Stagner AM, et al. Intraocular medulloepitheliomas and embryonal tumors with multilayered rosettes of the brain: comparative roles of LIN28A and C19MC. Am J Ophthalmol 2015; 159: 1065–74. ArticlePubMed
- 130. Cahill DP, Louis DN, Cairncross JG. Molecular background of oligodendroglioma: 1p/19q, IDH, TERT, CIC and FUBP1. CNS Oncol 2015; 4: 287–94. ArticlePubMedPMC
- 131. Jue TR, McDonald KL. The challenges associated with molecular targeted therapies for glioblastoma. J Neurooncol 2016; 127: 427–34. ArticlePubMedPDF
References
Figure & Data
References
Citations
- PTEN regulates expression of its pseudogene in glioblastoma cells in DNA methylation-dependent manner
Tatyana F. Kovalenko, Bhupender Yadav, Ksenia S. Anufrieva, Tatyana D. Larionova, Tatiana E. Aksinina, Yaroslav A. Latyshev, Soniya Bastola, Michail I. Shakhparonov, Amit Kumar Pandey, Marat S. Pavlyukov
Biochimie.2024; 219: 74. CrossRef - CDKN2A/B deletion in IDH-mutant astrocytomas: An evaluation by Fluorescence in-situ hybridization
Manali Ranade, Sridhar Epari, Omshree Shetty, Sandeep Dhanavade, Sheetal Chavan, Ayushi Sahay, Arpita Sahu, Prakash Shetty, Aliasgar Moiyadi, Vikash Singh, Archya Dasgupta, Abhishek Chatterjee, Sadhana Kannan, Tejpal Gupta
Journal of Neuro-Oncology.2024; 167(1): 189. CrossRef - Central neurocytoma exhibits radial glial cell signatures with FGFR3 hypomethylation and overexpression
Yeajina Lee, Tamrin Chowdhury, Sojin Kim, Hyeon Jong Yu, Kyung-Min Kim, Ho Kang, Min-Sung Kim, Jin Wook Kim, Yong-Hwy Kim, So Young Ji, Kihwan Hwang, Jung Ho Han, Jinha Hwang, Seong-Keun Yoo, Kyu Sang Lee, Gheeyoung Choe, Jae-Kyung Won, Sung-Hye Park, Yon
Experimental & Molecular Medicine.2024; 56(4): 975. CrossRef - Drugging Hijacked Kinase Pathways in Pediatric Oncology: Opportunities and Current Scenario
Marina Ferreira Candido, Mariana Medeiros, Luciana Chain Veronez, David Bastos, Karla Laissa Oliveira, Julia Alejandra Pezuk, Elvis Terci Valera, María Sol Brassesco
Pharmaceutics.2023; 15(2): 664. CrossRef - BrainBase: a curated knowledgebase for brain diseases
Lin Liu, Yang Zhang, Guangyi Niu, Qianpeng Li, Zhao Li, Tongtong Zhu, Changrui Feng, Xiaonan Liu, Yuansheng Zhang, Tianyi Xu, Ruru Chen, Xufei Teng, Rongqin Zhang, Dong Zou, Lina Ma, Zhang Zhang
Nucleic Acids Research.2022; 50(D1): D1131. CrossRef - A heat shock protein 90 inhibitor reduces oncoprotein expression and induces cell death in heterogeneous glioblastoma cells with EGFR, PDGFRA, CDK4, and NF1 aberrations
Kuan-Ta Ho, Pei-Fan Chen, Jian-Ying Chuang, Po-Wu Gean, Yuan-Shuo Hsueh
Life Sciences.2022; 288: 120176. CrossRef - Bicentric validation of the navigated transcranial magnetic stimulation motor risk stratification model
Tizian Rosenstock, Levin Häni, Ulrike Grittner, Nicolas Schlinkmann, Meltem Ivren, Heike Schneider, Andreas Raabe, Peter Vajkoczy, Kathleen Seidel, Thomas Picht
Journal of Neurosurgery.2022; 136(4): 1194. CrossRef - Foramen magnum meningioma presented as cervical myelopathy in a pregnant COVID-19 patient: A case report
Olivia Josephine Wijaya, Djohan Ardiansyah
Annals of Medicine and Surgery.2022; 77: 103647. CrossRef - The telomere maintenance mechanism spectrum and its dynamics in gliomas
Sojin Kim, Tamrin Chowdhury, Hyeon Jong Yu, Jee Ye Kahng, Chae Eun Lee, Seung Ah. Choi, Kyung-Min Kim, Ho Kang, Joo Ho Lee, Soon-Tae Lee, Jae-Kyung Won, Kyung Hyun Kim, Min-Sung Kim, Ji Yeoun Lee, Jin Wook Kim, Yong-Hwy Kim, Tae Min Kim, Seung Hong Choi,
Genome Medicine.2022;[Epub] CrossRef - A review of predictive, prognostic and diagnostic biomarkers for brain tumours: towards personalised and targeted cancer therapy
Ernest Osei, Pascale Walters, Olivia Masella, Quinton Tennant, Amber Fishwick, Eugenia Dadzie, Anmol Bhangu, Johnson Darko
Journal of Radiotherapy in Practice.2021; 20(1): 83. CrossRef - Use of a novel navigable tubular retractor system in 1826 minimally invasive parafascicular surgery (MIPS) cases involving deep-seated brain tumors, hemorrhages and malformations
Martina M. Cartwright, Penny Sekerak, Joseph Mark, Julian Bailes
Interdisciplinary Neurosurgery.2021; 23: 100919. CrossRef - Disentangling the therapeutic tactics in GBM: From bench to bedside and beyond
S. Daisy Precilla, Shreyas S. Kuduvalli, Anitha Thirugnanasambandhar Sivasubramanian
Cell Biology International.2021; 45(1): 18. CrossRef - Sequencing of a central nervous system tumor demonstrates cancer transmission in an organ transplant
Marie-Claude Gingras, Aniko Sabo, Maria Cardenas, Abbas Rana, Sadhna Dhingra, Qingchang Meng, Jianhong Hu, Donna M Muzny, Harshavardhan Doddapaneni, Lesette Perez, Viktoriya Korchina, Caitlin Nessner, Xiuping Liu, Hsu Chao, John Goss, Richard A Gibbs
Life Science Alliance.2021; 4(9): e202000941. CrossRef - Radiogenomics of Gliomas
Chaitra Badve, Sangam Kanekar
Radiologic Clinics of North America.2021; 59(3): 441. CrossRef - Chemical analysis of the human brain by imaging mass spectrometry
Akhila Ajith, Yeswanth Sthanikam, Shibdas Banerjee
The Analyst.2021; 146(18): 5451. CrossRef - Correlation between Stage and Histopathological Features and Clinical Outcomes in Patients with Glioma Tumors
Andre Lona, Alfansuri Kadri, Irina Kemala Nasution
Open Access Macedonian Journal of Medical Sciences.2021; 9(T3): 262. CrossRef - Pediatric Glioma: An Update of Diagnosis, Biology, and Treatment
Yusuke Funakoshi, Nobuhiro Hata, Daisuke Kuga, Ryusuke Hatae, Yuhei Sangatsuda, Yutaka Fujioka, Kosuke Takigawa, Masahiro Mizoguchi
Cancers.2021; 13(4): 758. CrossRef - Discovery of clinically relevant fusions in pediatric cancer
Stephanie LaHaye, James R. Fitch, Kyle J. Voytovich, Adam C. Herman, Benjamin J. Kelly, Grant E. Lammi, Jeremy A. Arbesfeld, Saranga Wijeratne, Samuel J. Franklin, Kathleen M. Schieffer, Natalie Bir, Sean D. McGrath, Anthony R. Miller, Amy Wetzel, Katheri
BMC Genomics.2021;[Epub] CrossRef - Clinicopathological correlation of glioma patients with respect to immunohistochemistry markers: A prospective study of 115 patients in a Tertiary Care Hospital in North India
Gitanshu Dahuja, Ashok Gupta, Arpita Jindal, Gaurav Jain, Santosh Sharma, Arvind Kumar
Asian Journal of Neurosurgery.2021; 16(04): 732. CrossRef - Role of Extracellular Vesicles in Glioma Progression: Deciphering Cellular Biological Processes to Clinical Applications
Rashmi Rana, Shikha Joon, Kirti Chauhan, Vaishnavi Rathi, Nirmal Kumar Ganguly, Chandni Kumari, Dharmendra Kumar Yadav
Current Topics in Medicinal Chemistry.2021; 21(8): 696. CrossRef - A comprehensive overview on the molecular biology of human glioma: what the clinician needs to know
P. D. Delgado-López, P. Saiz-López, R. Gargini, E. Sola-Vendrell, S. Tejada
Clinical and Translational Oncology.2020; 22(11): 1909. CrossRef - Use of Fluorescence In Situ Hybridization (FISH) in Diagnosis and Tailored Therapies in Solid Tumors
Natalia Magdalena Chrzanowska, Janusz Kowalewski, Marzena Anna Lewandowska
Molecules.2020; 25(8): 1864. CrossRef - From Banding to BAM Files
Adrian M. Dubuc
Surgical Pathology Clinics.2020; 13(2): 343. CrossRef - Mutation profiling of anaplastic ependymoma grade III by Ion Proton next generation DNA sequencing
Ejaz Butt, Sabra Alyami, Tahani Nageeti, Muhammad Saeed, Khalid AlQuthami, Abdellatif Bouazzaoui, Mohammad Athar, Zainularifeen Abduljaleel, Faisal Al-Allaf, Mohiuddin Taher
F1000Research.2020; 8: 613. CrossRef - Assessment of the non-linear optical behavior of cells for discrimination between normal and malignant glial cells
Soraya Emamgholizadeh Minaei, Alireza Ghader, Ali Abbasian Ardakani, Samideh Khoei, Mohammad Hosein Majles Ara
Laser Physics.2020; 30(12): 125601. CrossRef - Mutation profiling of anaplastic ependymoma grade III by Ion Proton next generation DNA sequencing
Muhammad Butt, Sabra Alyami, Tahani Nageeti, Muhammad Saeed, Khalid AlQuthami, Abdellatif Bouazzaoui, Mohammad Athar, Zainularifeen Abduljaleel, Faisal Al-Allaf, Mohiuddin Taher
F1000Research.2019; 8: 613. CrossRef - A Multiplex Quantitative Reverse Transcription Polymerase Chain Reaction Assay for the Detection of KIAA1549–BRAF Fusion Transcripts in Formalin-Fixed Paraffin-Embedded Pilocytic Astrocytomas
David Bret, Valentin Chappuis, Delphine Poncet, François Ducray, Karen Silva, Fabrice Mion, Alexandre Vasiljevic, Carole Ferraro-Peyret, Carmine Mottolese, Pierre Leblond, Mathieu Gabut, Didier Frappaz, Nathalie Streichenberger, David Meyronet, Pierre-Pau
Molecular Diagnosis & Therapy.2019; 23(4): 537. CrossRef - A PTPmu Biomarker is Associated with Increased Survival in Gliomas
Mette L. Johansen, Jason Vincent, Haley Gittleman, Sonya E. L. Craig, Marta Couce, Andrew E. Sloan, Jill S. Barnholtz-Sloan, Susann M. Brady-Kalnay
International Journal of Molecular Sciences.2019; 20(10): 2372. CrossRef -
ATRX Mutations in Pineal Parenchymal Tumors of Intermediate Differentiation
Haydee Martínez, Michelle Nagurney, Zi-Xuan Wang, Charles G Eberhart, Christopher M Heaphy, Mark T Curtis, Fausto J Rodriguez
Journal of Neuropathology & Experimental Neurology.2019; 78(8): 703. CrossRef - The role of fibroblast growth factors and their receptors in gliomas: the mutations involved
Vasiliki Georgiou, Vasiliki Gkretsi
Reviews in the Neurosciences.2019; 30(5): 543. CrossRef - The Role of Next Generation Sequencing in Diagnosis of Brain Tumors: A Review Study
Sadegh Shirian, Yahya Daneshbod, Saranaz Jangjoo, Amir Ghaemi, Arash Goodarzi, Maryam Ghavideldarestani, Ahmad Emadi, Arman Ai, Akbar Ahmadi, Jafar Ai
Archives of Neuroscience.2019;[Epub] CrossRef - Applied Precision Cancer Medicine in Neuro-Oncology
H. Taghizadeh, L. Müllauer, J. Furtner, J. A. Hainfellner, C. Marosi, M. Preusser, G. W. Prager
Scientific Reports.2019;[Epub] CrossRef - Comparative genomic analysis of driver mutations in matched primary and recurrent meningiomas
Joshua Loewenstern, John Rutland, Corey Gill, Hanane Arib, Margaret Pain, Melissa Umphlett, Yayoi Kinoshita, Russell McBride, Michael Donovan, Robert Sebra, Joshua Bederson, Mary Fowkes, Raj Shrivastava
Oncotarget.2019; 10(37): 3506. CrossRef - Next generation DNA sequencing of atypical choroid plexus papilloma of brain: Identification of novel mutations in a female patient by Ion Proton
Mohiuddin Taher, Amal Hassan, Muhammad Saeed, Raid Jastania, Tahani Nageeti, Hisham Alkhalidi, Ghida Dairi, Zainularifeen Abduljaleel, Mohammad Athar, Abdellatif Bouazzaoui, Wafa El‑Bjeirami, Faisal Al‑Allaf
Oncology Letters.2019;[Epub] CrossRef - Pure mechanistic analysis of additive neuroprotective effects between baicalin and jasminoidin in ischemic stroke mice
Peng-qian Wang, Qiong Liu, Wen-juan Xu, Ya-nan Yu, Ying-ying Zhang, Bing Li, Jun Liu, Zhong Wang
Acta Pharmacologica Sinica.2018; 39(6): 961. CrossRef - The Smad4/PTEN Expression Pattern Predicts Clinical Outcomes in Colorectal Adenocarcinoma
Yumin Chung, Young Chan Wi, Yeseul Kim, Seong Sik Bang, Jung-Ho Yang, Kiseok Jang, Kyueng-Whan Min, Seung Sam Paik
Journal of Pathology and Translational Medicine.2018; 52(1): 37. CrossRef - Primary brain tumours in adults
Sarah Lapointe, Arie Perry, Nicholas A Butowski
The Lancet.2018; 392(10145): 432. CrossRef - Epidemiology and Overview of Gliomas
Mary Elizabeth Davis
Seminars in Oncology Nursing.2018; 34(5): 420. CrossRef - Toward Precision Medicine: Promising Areas of Research in Glioma
Mary Elizabeth Davis, Dana Bossert, Malbora Manne
Seminars in Oncology Nursing.2018; 34(5): 569. CrossRef - Molecular Basis of Pediatric Brain Tumors
Alexia Klonou, Christina Piperi, Antonios N. Gargalionis, Athanasios G. Papavassiliou
NeuroMolecular Medicine.2017; 19(2-3): 256. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
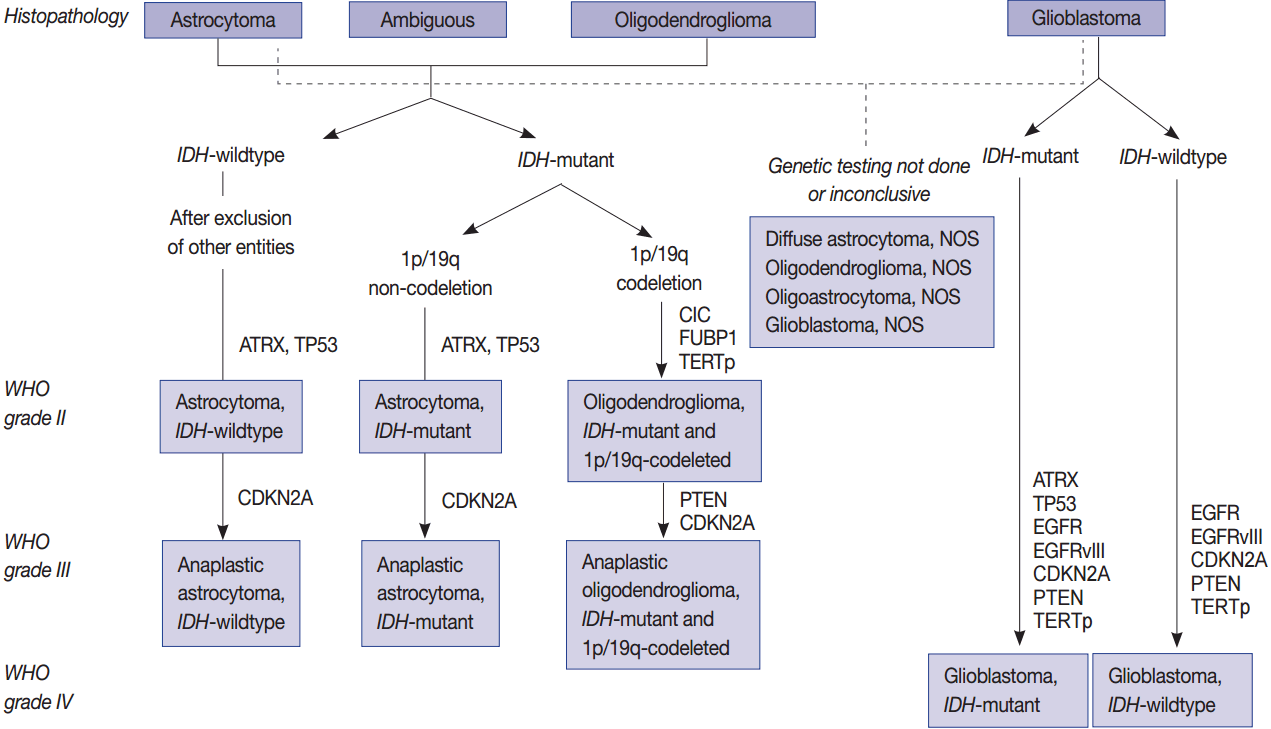
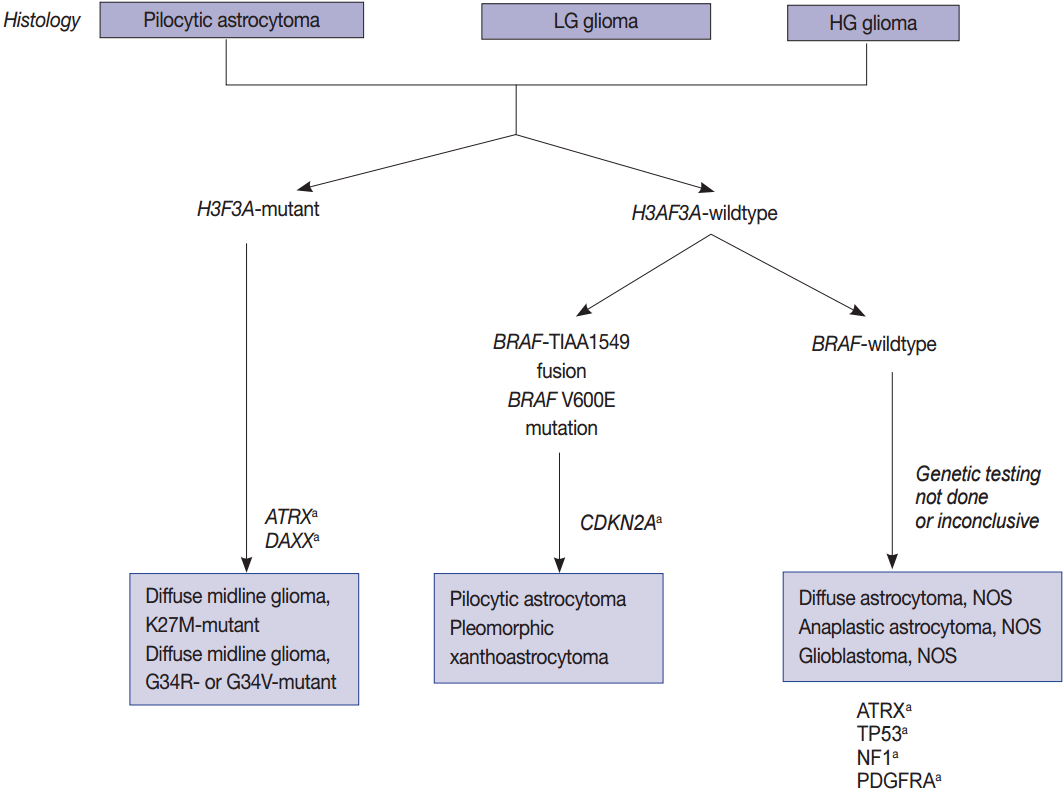
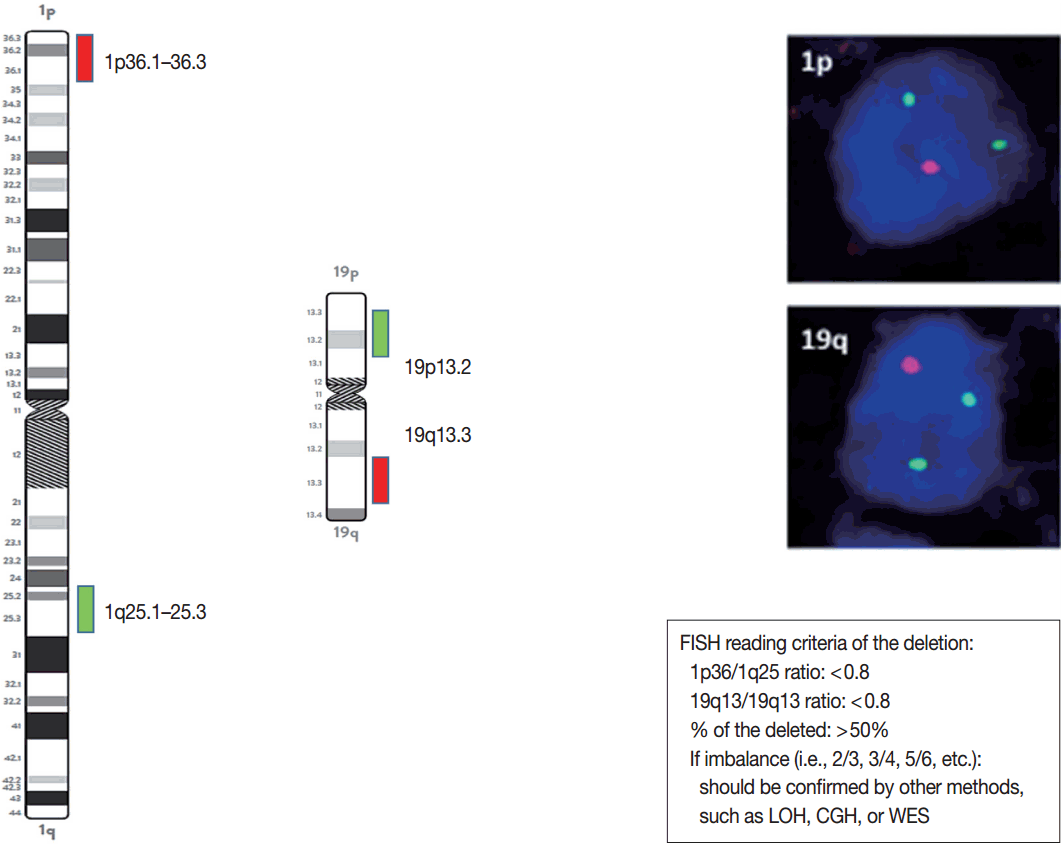
- Related articles

