E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 46(1); 2012 > Article
-
Review
CpG Island Hypermethylation in Gastric Carcinoma and Its Premalignant Lesions - Gyeong Hoon Kang
-
Korean Journal of Pathology 2012;46(1):1-9.
DOI: https://doi.org/10.4132/KoreanJPathol.2012.46.1.1
Published online: February 23, 2012
Department of Pathology, Cancer Research Institute, Seoul National University College of Medicine, Seoul, Korea.
- Corresponding Author: Gyeong Hoon Kang, M.D. Department of Pathology, Cancer Research Institute, Seoul National University College of Medicine, 28 Yeongeon-dong, Jongno-gu, Seoul 110-744, Korea. Tel: +82-2-740-8263, Fax: +82-2-743-5530, 'ghkang@snu.ac.kr'
• Received: November 4, 2011 • Revised: November 15, 2011 • Accepted: November 21, 2011
© 2012 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Gastric cancers arise through a multistep process characterized by the progressive accumulation of molecular alterations in which genetic and epigenetic mechanisms have been implicated. Gastric cancer is one of the human malignancies in which aberrant promoter CpG island hypermethylation is frequently found. Helicobacter pylori and Epstein-Barr virus, which are known carcinogens for gastric cancer, are closely associated with enhanced hypermethylation of CpG island loci in gastric non-neoplastic epithelial cells and cancer cells, respectively. Aberrant CpG island hypermethylation occurs early in the multistep cascade of gastric carcinogenesis and tends to increase with the step-wise progression of the lesion. Approximately 400 genes that are actively expressed in normal gastric epithelial cells are estimated to be inactivated in gastric cancers as a result of promoter CpG island hypermethylation. In this review, a variety of information is summarized regarding CpG island hypermethylation in gastric cancer.
- How many genes are known to be methylated in gastric cancer?
- In 1996, Lee et al.5 reported for the first time that p16 was inactivated in gastric cancer by promoter CpG island hypermethylation rather than by genetic mutation. Since then, many researchers have demonstrated cancer-specific hypermethylation and inactivation of candidate genes in gastric cancer tissues and have correlated this hypermethylation with clinicopathologic features. However, the number of genes demonstrated to be inactivated by promoter CpG island hypermethylation was limited until the application of array-based genome-scale DNA methylation analysis for gastric cancers. With the application of 5-aza-2'-deoxycytidine (DAC) treatment and oligonucleotide microarrays, Yamashita et al.6 estimated that approximately 421 genes are silenced by promoter CpG island hypermethylation in a gastric cancer cell line (AGS). Considering the fact that promoter CpG island hypermethylation is exaggerated in a cancer cell line compared with primary cancer tissue and that the AGS cell line has an increased rate of de novo methylation because of over-expression of DNMT3b, the actual number of genes silenced by promoter CpG island hypermethylation can be estimated to be less than 421 in primary gastric cancers. Our team also performed bead array-based expression analysis of gastric cancer cell lines before and after DAC treatment with subsequent confirmation of CpG island hypermethylation of the candidate genes by methylation-specific PCR. We found 140 novel genes that are silenced by promoter CpG island hypermethylation in primary gastric cancer tissue (Jung et al. in preparation).
- The timing of hypermethylation in multistep gastric carcinogenesis
- Promoter CpG island hypermethylation is now recognized to be an important mechanism responsible for the inactivation of tumor suppressor genes or tumor-related genes. If promoter CpG island hypermethylation of some genes plays an important role in the malignant transformation of gastric epithelial cells, this pattern of hypermethylation should be found in premalignant lesions of the stomach, including gastric adenomas and intestinal metaplasia. In order to determine the frequency and timing of hypermethylation during multistep gastric carcinogenesis, Kang et al. analyzed multistep lesions of the stomach, in regards to their methylation status, in five genes7 or 12 genes8 using methylation-specific PCR or in 25 genes using MethyLight analysis;9 they demonstrated that promoter CpG island hypermethylation occurs early in multistep gastric carcinogenesis and accumulates during progression of the gastric lesion along the multistep carcinogenesis pathway. During multistep gastric carcinogenesis, there is a steep rise in the number of methylated genes when progressing from chronic gastritis to intestinal metaplasia, which was a consistent finding in a series of studies.7-9 Regardless of the status of Helicobacter pylori infection, the number of methylated genes in intestinal metaplasia was significantly higher than that found in chronic gastritis without intestinal metaplasia.9 This suggests that intestinal metaplasia is an epigenetically altered lesion. However, even in chronic gastritis without intestinal metaplasia, promoter CpG island hypermethylation occurs in association with H. pylori infection10 and aging.11,12
- Helicobacter pylori infection-associated DNA hypermethylation
- H. pylori has been designated as a human class I carcinogen for gastric malignancy by the International Agency for Research on Cancer. Although the exact mechanism of H. pylori-associated gastric carcinogenesis is unknown, long-standing bacterial infection, perpetuated chronic inflammation, and sustained mucosal epithelial cell proliferation are thought to produce a carcinogenic environment. Chan et al.10 were the first to demonstrate H. pylori-associated hypermethylation in the gastric epithelia, which was supported by subsequent studies demonstrating that the eradication of H. pylori infection results in a reversal of the methylation status of multiple CpG island loci.13-15 Thus, it is plausible that aberrant methylation induced by H. pylori infection may contribute to H. pylori infection-associated gastric carcinogenesis. It has been reported that interleukin 1 beta can modulate CpG island methylation through the activation of DNA methyltransferase.16 In an in vitro study, interleukin 1 beta siRNA blocked H. pylori-induced methylation of the CDH1 promoter CpG island locus in a gastric cancer cell line.17 In an animal model experiment by the Ushijima team, H. pylori infection resulted in the induction of CpG island hypermethylation of candidate genes, and the eradication led to marked decreases in methylation levels in the candidate genes. However, the suppression of inflammation by treatment with the immunosuppressive drug cyclosporine blocked the induction of DNA methylation in the candidate genes. These findings suggest that the infection-associated inflammatory response, rather than H. pylori itself, was responsible for the induction of altered DNA methylation.18 In a subsequent gerbil study, neutrophilic inflammation caused by treatment with ethanol or NaCl did not induce DNA methylation in candidate genes, whereas chronic inflammation caused by H. pylori or H. felis infection led to altered methylation in candidate genes. This finding suggests that it is not the inflammation itself, but rather specific types of inflammation, that are necessary for methylation induction.19
- Aging-related hypermethylation vs inflammation-related hypermethylation
- Challenging traditional thought regarding the lack of CpG island methylation in normal tissues, a recent study indicated that 4-8% of CpG island loci are methylated in the genomic DNA of human blood, brain, muscle, and spleen tissue.20,21 Additionally, normal cells have been shown to acquire hypermethylation in an aging-related manner: aging-related methylation was first demonstrated for the oestrogen receptor (ER) gene by Issa et al.22 and has subsequently been demonstrated in multiple genes by Ahuja et al.23 In the stomach, Waki et al.11 reported aging-related methylation of CDH1, MLH1, and p16 in non-neoplastic gastric epithelia. However, because CpG island hypermethylation can be induced by chronic inflammation in the stomach and because the prevalence of H. pylori infection increases with age,24 the interplay between aging and chronic inflammation is complicated by H. pylori infection. Chan et al.25 found that CDH1 methylation was associated with age in the stomach, the presence of chronic gastritis, and H. pylori infection using a univariate analysis, but H. pylori infection was the only independent factor associated with CDH1 methylation in the multivariate analysis. Furthermore, Maekita et al.26 reported that there was no aging-related hypermethylation in healthy gastric mucosa. However, a current study by our team supports the presence of aging-related hypermethylation in the gastric mucosa because the number of methylated genes was significantly higher in H. pylori-negative adult stomach samples than in H. pylori-negative pediatric samples.27 In WI-38 human embryonic lung fibroblasts, p16 was found to undergo spontaneous promoter CpG island hypermethylation during passage of these cells in culture,28 which supports the presence of age-related hypermethylation. Thus, we cannot exclude the possibility that aging-related methylation occurs in non-neoplastic gastric epithelia without H. pylori infection.
- Field cancerization
- Many studies have shown that methylation levels or frequencies of multiple genes are higher in gastric mucosa from gastric cancer patients than in mucosa from non-cancer subjects.11,26,29 In a stomach with enhanced CpG island hypermethylation, the affected cells may have a growth-selective advantage imparted by the expressional loss of the methylated genes, which may predispose the cells to acquiring further genetic or epigenetic defects that lead to neoplasia. Synchronous multiple gastric cancers, constituting 4-9% of gastric cancers, occur at an older age and are more commonly associated with an extensive distribution of intestinal metaplasia in the background mucosa compared with single gastric cancer.30,31 In a recent study, non-neoplastic gastric mucosa from synchronous gastric cancer patients was found to have more hypermethylated genes than non-neoplastic gastric mucosa from patients with a single gastric cancer, which suggests that enhanced hypermethylation in the background gastric mucosa might contribute to the development of multiple gastric cancers.32 Multiple gastric cancers are not only a genetic model but also an epigenetic model for field cancerization. Based on the findings that methylation levels in gastric mucosa are significantly increased in cases with a single gastric cancer and even more so in cases with multiple gastric cancers, it has been suggested that quantitative information regarding methylation levels of specific genes may serve as a biomarker for predicting an individual's risk for developing gastric cancer.32,33 However, it should be noted that increased methylation levels or frequencies in gastric mucosa from gastric cancer patients may reflect a higher prevalence of H. pylori infection and intestinal metaplasia (compared with gastric mucosa from non-cancer subjects).34,35 Because both H. pylori infection and intestinal metaplasia are closely linked with increased CpG island hypermethylation, further clarification is needed regarding whether non-neoplastic gastric mucosa without intestinal metaplasia from H. pylori-negative gastric cancer patients harbors more hypermethylated genes than non-neoplastic gastric mucosa from H. pylori-negative non-cancer subjects. In Park et al.'s study,9 no significant differences were observed in the number of methylated genes or methylation levels of 25 individual genes in chronic gastritis tissue from cancer and non-cancer patients after normalization of the confounding factors of H. pylori and intestinal metaplasia. Because synchronous multiple gastric cancers tend to be associated with widespread intestinal metaplasia when compared with single gastric cancers, cancer-associated gastric mucosa might show higher methylation frequencies or higher methylation levels of multiple CpG island loci in multiple synchronous gastric cancer patients than in single gastric cancer patients.
- Prognostic implications of individual gene methylation
- Promoter CpG island hypermethylation can be utilized as a tumor biomarker for the detection of tumor cells in gastric juice or serum or for the prediction of clinical outcomes. A dozen DNA methylation markers have been reported to be closely associated with worse or better clinical outcomes in gastric cancer patients; MAL or COX2 methylation was correlated with better clinical outcomes in gastric cancer patients,36,37 whereas DAPK, TMS1, IQGAP2, SOX2, CACNA2D3, DKK-3, TFPI2, and Cystatin methylation has been correlated with worse clinical outcomes in gastric cancer patients.38-44 However, most of the studies that have evaluated these DNA methylation markers for their prognostic implication used methylation-specific polymerase chain reaction (MSP). Although MSP is a highly sensitive method for detecting one methylated allele in 10,000 unmethylated alleles,45 it is unreliable for detecting low levels of methylation. Another issue related to these DNA methylation markers is that a validation study was not performed to prove their utility as a prognostic marker. The final issue concerns concordance among individual gene hypermethylation: hypermethylation of one specific gene tends to be concordant with that of another individual gene; thus, better or worse survival observed in gastric cancers with hypermethylation of an individual gene may not be attributed to hypermethylation of that particular gene. Rather, survival may be related to concordant hypermethylation of multiple CpG island loci, namely CpG island methylator phenotype (CIMP).
- CIMP-positive gastric cancer
- CIMP refers to a subset of malignancies that is characterized by widespread hypermethylation of multiple promoter CpG island loci. Since the CIMP concept was first introduced for the molecular pathways of colorectal cancers (CRCs) by Dr. Issa's group,46 many investigators have attempted to characterize the clinicopathological and molecular features of CIMP-positive CRCs and have found a close association with proximal colon location, older age at onset, poor differentiation, microsatellite instability (MSI), and BRAF mutations.47-49 Similarly, the presence of CIMP-positive gastric cancers has been reported by the same group,50 which did not find distinct clinicopathologic features of CIMP-positive gastric cancers, with the exception of a close association with MSI. Since then, several researchers have attempted to characterize the clinicopathological features of CIMP-positive gastric cancers using variable methodologies for DNA methylation analysis and their own CIMP marker panels; this has led to controversial results.51-56 Despite variable findings, relatively common findings include close associations of CIMP-positive gastric cancers with diffuse type-histology and better clinical outcomes. Recently, with the exclusion of MSI-positive gastric cancers and Epstein-Barr virus (EBV)-positive gastric cancers from the analysis, Park et al.57 found characteristic clinicopathologic features of CIMP-positive gastric cancers that were defined as tumors with methylation of 13 or more markers during the analysis of the 16 cancer-specific DNA methylation markers; these defined CIMP-positive gastric cancers tended to show distinct clinicopathologic features, including a proclivity toward diffuse or mixed-type histology, poor differentiation, infiltrative gross types, and higher cancer stages.
- EBV-associated gastric cancer
- In addition to H. pylori, EBV has also been recognized as a gastric cancer-causing infectious agent.58,59 EBV-associated gastric cancer, comprising nearly 10% of gastric cancers, are characterized by a younger age at onset, male predominance, proximal location, frequent association of lymphoid stroma, and a better prognosis.60 EBV-associated gastric cancer is a prototype of the CIMP-positive types of gastric cancer and exhibits consistently higher frequencies and levels of methylation in examined cancer-related methylation markers,53,57,61 which is independent of whether the EBV-associated gastric cancer is histologically lymphoepithelioma-like or ordinary.60 Because the number of methylated cancer-specific methylation markers is far higher in EBV-associated gastric cancers than in EBV-negative gastric cancers, EBV-associated aberrant hypermethylation is a global event. However, MLH1 methylation and resultant MSI are, if ever, rarely observed in EBV-associated gastric cancers. Although it can be speculated that the methylation machinery of EBV-associated gastric cancer is capable of recognizing its own methylation targets, it may be hypothesized that the presence of both CIMP and MSI could cause a growth disadvantage and subsequently lead to the negative selection of gastric cancer cells containing both CIMP and MSI. Tumor cells from EBV-associated gastric cancers display DNA methyltransferase I over-expression,62 which is closely associated with interleukin-1-beta (IL1B) over-expression60 or latent membrane protein 2A-associated phosphorylation of signal transducer and activator of transcription 3 (STAT3) in EBV-associated gastric cancers.63 IL1B is capable of increasing the expression of DNMT1 via the production of nitric oxide, and phosphorylated STAT3 binds to the DNMT1 promoter and induces transcription.
- MSI-positive gastric cancer
- The frequency of MSI-positive gastric cancer varies from 8% to 37%. In gastric cancer, MSI is mainly caused by promoter CpG island hypermethylation of the MLH1 gene. Somatic mutations of mismatch repair genes are very rare in sporadic gastric cancers. Thus, known clinicopathological features of MSI-positive gastric cancer, including female sex, older age of onset, antral location, ulcerofungating gross type by Borrmann's classification, intestinal type by Lauren classification, expanding type by Ming classification, and better survival64 represent features of sporadic MSI-positive gastric cancer. Since gastric cancer is an extracolonic lesion in Lynch syndrome, we have encountered MSI-positive gastric cancer without MLH1 methylation. Recently, our team has compared clinicopathological features between MSI-positive gastric cancers with and without MLH1 methylation (Kim et al. in preparation). Of the known clinicopathological features for MSI-positive gastric cancer, female preponderance, older age of onset and antral location do not correspond with Lynch syndrome-associated MSI-positive gastric cancer.65 Several studies have shown that MLH1 methylation occurs in premalignant stages, including intestinal metaplasia and gastric adenoma. Thus, MLH1 methylation has been considered an early event during multistep gastric carcinogenesis.7,9,66,67 However, a recent study of Ling et al.68 has shown that MSI can develop from MSI-low or the absence of MSI due to time-dependent accumulation of DNA methylation during progression of early stage-gastric cancer, which indicates that silencing of MLH1 due to promoter hypermethylation may appear as a later event during multistep gastric carcinogenesis. Unfortunately, the study did not investigate whether later acquisition of MLH1 methylation occurs as a phenomenon of CIMP. MLH1 methylation is unlikely to occur as an isolated sporadic event without concurrent hypermethylation of multiple gene promoter CpG island loci.
- Epigenetic regulation of microRNA
- A new class of small non-coding RNAs known as microRNAs (miRNA) have recently been discovered. Mature miRNAs, 21 to 30 nucleotide-sized, are cleaved from 70- to 100-nucleotide hairpin miRNA precursors in the cytoplasm by the RNase III enzyme Dicer. Base-pairing between the miRNA strand and the 3' untranslated regions of its target mRNAs (potentially hundreds of genes) directs RNA-induced silencing complex to either cause mRNA degradation or translational repression. Through these molecular mechanisms, miRNAs serve as key regulators of gene expression involved in crucial cellular processes, including development, proliferation, cellular differentiation, and apoptosis. Although the generation of miRNAs and their mode of action in regulating gene function have been intensively studied, the regulation of miRNA expression remains largely unclear. The means by which miRNA is regulated is somewhat complicated. Recently, Saito et al. demonstrated for the first time that miR-127 is regulated by DNA hypermethylation69 and since then, a dramatically increased number of studies have documented the epigenetic regulation of miRNAs.70 However, in gastric cancer, approximately 113 miRNAs have been demonstrated to be dysregulated compared with normal gastric mucosal tissues but a dozen miRNA genes have been shown to be downregulated by aberrant hypermethylation, including miR-512-5p,71 miR-375,72 miR-212,73 miR-181c,73 miR-196b,74 miR-137,75 miR-129-2,76 miR-124a-1, miR-124a-2, miR-124a-3,77 miR-34b, and miR-34c.78 In particular, Ando et al.77 demonstrated that miR-124a-1, -2, and -3 are frequently methylated in primary gastric cancer and in normal gastric mucosal tissues from healthy individuals with H. pylori infections. Among H. pylori-negative individuals, methylation levels are significantly higher in non-cancerous gastric mucosal tissues from gastric cancer patients than gastric mucosal tissues from healthy individuals, which suggest that methylation of miRNA genes comprises a field defect contributing to the pathogenesis of gastric cancer.77 Suzuki et al.78 also reported that miR-34b/c methylation is significantly associated with H. pylori infection among healthy individuals.
- Future perspective
- Although it is well known that EBV-positive gastric cancer is featured with extensive hypermethylation of multiple genes, the mechanism leading to genome-wide hypermethylation is still unknown. Because EBV-positive gastric dysplasia or adenoma has never been reported, it seems likely that EBV-infected epithelial cells transform directly into malignant cells. DNMT1 elevation in association with EBV infection does not provide a satisfactory explanation for genome-wide extensive hypermethylation because in vitro transfection of DNMT1 in cell lines or DNMT1 elevation in association with H. pylori infection also does not lead to such extensive hypermethylation as seen in EBV-positive gastric cancer. If we can elucidate the mechanism of EBV-associated extensive hypermethylation, we will have a better understanding of how promoter CpG island hypermethylation occurs.
- Although gastric cancer has shown a higher number of genes methylated compared with colorectal cancer, clinicopathological features of CIMP-positive gastric cancer are still obscure and marker panels diagnosing CIMP-positive gastric cancer are not established yet, which is in contrast to the situation in colorectal cancer. It is imperative to develop CIMP panel markers enabling the diagnosis of CIMP-positive gastric cancer and then to characterize clinicopathological features of CIMP-positive gastric cancer. In case these are accomplished, we expect to identify the precursor lesions of CIMP-positive gastric cancer and to delineate multistep morphological progression of CIMP-positive tumors.
GASTRIC CANCER METHYLATION CHANGE
- Although molecular pathways and morphological pathways are not well established for gastric cancers when compared with colorectal cancers, rapid development of methylation analysis technology will enable us to take a glimpse at the landscape of epigenetic alterations occurring at each step of multistep gastric carcinogenesis, which will provide molecular insights on morphological progression pathways. In addition, epigenetic studies may offer great potential for the identification of tumor biomarkers that can be utilized to detect and diagnose gastric cancer at its earliest stages, to accurately assess an individual's risk for gastric cancer, or to predict the response to chemotherapy or clinical outcomes.
CONCLUSION
Acknowledgments
Acknowledgments
- 1. Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A 2006; 103: 1412–1417. PMID: 16432200. ArticlePubMedPMC
- 2. Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res 2001; 61: 3225–3229. PMID: 11309270. PubMed
- 3. Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 1998; 72: 141–196. PMID: 9338076. ArticlePubMed
- 4. Park SY, Kim BH, Kim JH, et al. Methylation profiles of CpG island loci in major types of human cancers. J Korean Med Sci 2007; 22: 311–317. PMID: 17449942. ArticlePubMedPMC
- 5. Lee YY, Kang SH, Seo JY, et al. Alterations of p16INK4A and p15INK4B genes in gastric carcinomas. Cancer 1997; 80: 1889–1896. PMID: 9366289. ArticlePubMed
- 6. Yamashita S, Tsujino Y, Moriguchi K, Tatematsu M, Ushijima T. Chemical genomic screening for methylation-silenced genes in gastric cancer cell lines using 5-aza-2'-deoxycytidine treatment and oligonucleotide microarray. Cancer Sci 2006; 97: 64–71. PMID: 16367923. ArticlePubMed
- 7. Kang GH, Shim YH, Jung HY, Kim WH, Ro JY, Rhyu MG. CpG island methylation in premalignant stages of gastric carcinoma. Cancer Res 2001; 61: 2847–2851. PMID: 11306456. PubMed
- 8. Kang GH, Lee S, Kim JS, Jung HY. Profile of aberrant CpG island methylation along multistep gastric carcinogenesis. Lab Invest 2003; 83: 519–526. PMID: 12695555. ArticlePubMed
- 9. Park SY, Yoo EJ, Cho NY, Kim N, Kang GH. Comparison of CpG island hypermethylation and repetitive DNA hypomethylation in premalignant stages of gastric cancer, stratified for Helicobacter pylori infection. J Pathol 2009; 219: 410–416. PMID: 19639607. ArticlePubMed
- 10. Chan AO, Lam SK, Wong BC, et al. Promoter methylation of E-cadherin gene in gastric mucosa associated with Helicobacter pylori infection and in gastric cancer. Gut 2003; 52: 502–506. PMID: 12631658. ArticlePubMedPMC
- 11. Waki T, Tamura G, Tsuchiya T, Sato K, Nishizuka S, Motoyama T. Promoter methylation status of E-cadherin, hMLH1, and p16 genes in nonneoplastic gastric epithelia. Am J Pathol 2002; 161: 399–403. PMID: 12163364. ArticlePubMedPMC
- 12. Kang GH, Lee HJ, Hwang KS, Lee S, Kim JH, Kim JS. Aberrant CpG island hypermethylation of chronic gastritis, in relation to aging, gender, intestinal metaplasia, and chronic inflammation. Am J Pathol 2003; 163: 1551–1556. PMID: 14507661. ArticlePubMedPMC
- 13. Chan AO, Peng JZ, Lam SK, et al. Eradication of Helicobacter pylori infection reverses E-cadherin promoter hypermethylation. Gut 2006; 55: 463–468. PMID: 16428266. ArticlePubMedPMC
- 14. Leung WK, Man EP, Yu J, et al. Effects of Helicobacter pylori eradication on methylation status of E-cadherin gene in noncancerous stomach. Clin Cancer Res 2006; 12: 3216–3221. PMID: 16707623. ArticlePubMed
- 15. Ushijima T, Nakajima T, Maekita T. DNA methylation as a marker for the past and future. J Gastroenterol 2006; 41: 401–407. PMID: 16799880. ArticlePubMed
- 16. Hmadcha A, Bedoya FJ, Sobrino F, Pintado E. Methylation-dependent gene silencing induced by interleukin 1beta via nitric oxide production. J Exp Med 1999; 190: 1595–1604. PMID: 10587350. ArticlePubMedPMC
- 17. Qian X, Huang C, Cho CH, Hui WM, Rashid A, Chan AO. E-cadherin promoter hypermethylation induced by interleukin-1beta treatment or H. pylori infection in human gastric cancer cell lines. Cancer Lett 2008; 263: 107–113. PMID: 18249489. ArticlePubMed
- 18. Niwa T, Tsukamoto T, Toyoda T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res 2010; 70: 1430–1440. PMID: 20124475. ArticlePubMed
- 19. Hur K, Niwa T, Toyoda T, et al. Insufficient role of cell proliferation in aberrant DNA methylation induction and involvement of specific types of inflammation. Carcinogenesis 2011; 32: 35–41. PMID: 20980348. ArticlePubMed
- 20. Illingworth R, Kerr A, Desousa D, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol 2008; 6: e22PMID: 18232738. ArticlePubMedPMC
- 21. Shen L, Kondo Y, Guo Y, et al. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet 2007; 3: 2023–2036. PMID: 17967063. ArticlePubMedPMC
- 22. Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet 1994; 7: 536–540. PMID: 7951326. ArticlePubMed
- 23. Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998; 58: 5489–5494. PMID: 9850084. PubMed
- 24. Drumm B, Perez-Perez GI, Blaser MJ, Sherman PM. Intrafamilial clustering of Helicobacter pylori infection. N Engl J Med 1990; 322: 359–363. PMID: 2300088. ArticlePubMed
- 25. Chan AO, Lam SK, Wong BC, Kwong YL, Rashid A. Gene methylation in non-neoplastic mucosa of gastric cancer: age or Helicobacter pylori related? Am J Pathol 2003; 163: 370–371. PMID: 12819044. ArticlePubMedPMC
- 26. Maekita T, Nakazawa K, Mihara M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res 2006; 12(3 Pt 1):989–995. PMID: 16467114. ArticlePubMed
- 27. Shin SH, Park SY, Ko JS, Kim N, Kang GH. Aberrant CpG island hypermethylation in pediatric gastric mucosa in association with Helicobacter pylori infection. Arch Pathol Lab Med 2011; 135: 759–765. PMID: 21631269. ArticlePubMed
- 28. Barzily-Rokni M, Friedman N, Ron-Bigger S, Isaac S, Michlin D, Eden A. Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A). Nucleic Acids Res 2011; 39: 1326–1335. PMID: 21030442. ArticlePubMedPMC
- 29. Leung WK, Yu J, Ng EK, et al. Concurrent hypermethylation of multiple tumor-related genes in gastric carcinoma and adjacent normal tissues. Cancer 2001; 91: 2294–2301. PMID: 11413518. ArticlePubMed
- 30. Honmyo U, Misumi A, Murakami A, Haga Y, Akagi M. Clinicopathological analysis of synchronous multiple gastric carcinoma. Eur J Surg Oncol 1989; 15: 316–321. PMID: 2547661. PubMed
- 31. Marrano D, Viti G, Grigioni W, Marra A. Synchronous and metachronous cancer of the stomach. Eur J Surg Oncol 1987; 13: 493–498. PMID: 3691822. PubMed
- 32. Nakajima T, Maekita T, Oda I, et al. Higher methylation levels in gastric mucosae significantly correlate with higher risk of gastric cancers. Cancer Epidemiol Biomarkers Prev 2006; 15: 2317–2321. PMID: 17119066. ArticlePubMed
- 33. Kaise M, Yamasaki T, Yonezawa J, Miwa J, Ohta Y, Tajiri H. CpG island hypermethylation of tumor-suppressor genes in H. pylori-infected non-neoplastic gastric mucosa is linked with gastric cancer risk. Helicobacter 2008; 13: 35–41. PMID: 18205664. Article
- 34. Stemmermann GN. Intestinal metaplasia of the stomach: a status report. Cancer 1994; 74: 556–564. PMID: 8033033. ArticlePubMed
- 35. Shimoyama T, Fukuda S, Tanaka M, Nakaji S, Munakata A. Evaluation of the applicability of the gastric carcinoma risk index for intestinal type cancer in Japanese patients infected with Helicobacter pylori. Virchows Arch 2000; 436: 585–587. PMID: 10917173. ArticlePubMed
- 36. Buffart TE, Overmeer RM, Steenbergen RD, et al. MAL promoter hypermethylation as a novel prognostic marker in gastric cancer. Br J Cancer 2008; 99: 1802–1807. PMID: 19002170. ArticlePubMedPMC
- 37. de Maat MF, van de Velde CJ, Umetani N, et al. Epigenetic silencing of cyclooxygenase-2 affects clinical outcome in gastric cancer. J Clin Oncol 2007; 25: 4887–4894. PMID: 17971584. ArticlePubMed
- 38. Kato K, Iida S, Uetake H, et al. Methylated TMS1 and DAPK genes predict prognosis and response to chemotherapy in gastric cancer. Int J Cancer 2008; 122: 603–608. PMID: 17943730. ArticlePubMed
- 39. Jin SH, Akiyama Y, Fukamachi H, Yanagihara K, Akashi T, Yuasa Y. IQGAP2 inactivation through aberrant promoter methylation and promotion of invasion in gastric cancer cells. Int J Cancer 2008; 122: 1040–1046. PMID: 17957782. ArticlePubMed
- 40. Otsubo T, Akiyama Y, Yanagihara K, Yuasa Y. SOX2 is frequently downregulated in gastric cancers and inhibits cell growth through cell-cycle arrest and apoptosis. Br J Cancer 2008; 98: 824–831. PMID: 18268498. ArticlePubMedPMC
- 41. Wanajo A, Sasaki A, Nagasaki H, et al. Methylation of the calcium channel-related gene, CACNA2D3, is frequent and a poor prognostic factor in gastric cancer. Gastroenterology 2008; 135: 580–590. PMID: 18588891. ArticlePubMed
- 42. Yu J, Tao Q, Cheng YY, et al. Promoter methylation of the Wnt/beta-catenin signaling antagonist Dkk-3 is associated with poor survival in gastric cancer. Cancer 2009; 115: 49–60. PMID: 19051296. ArticlePubMed
- 43. Jee CD, Kim MA, Jung EJ, Kim J, Kim WH. Identification of genes epigenetically silenced by CpG methylation in human gastric carcinoma. Eur J Cancer 2009; 45: 1282–1293. PMID: 19195878. ArticlePubMed
- 44. Chen X, Cao X, Dong W, et al. Cystatin M expression is reduced in gastric carcinoma and is associated with promoter hypermethylation. Biochem Biophys Res Commun 2010; 391: 1070–1074. PMID: 20004178. ArticlePubMed
- 45. Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93: 9821–9826. PMID: 8790415. ArticlePubMedPMC
- 46. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999; 96: 8681–8686. PMID: 10411935. ArticlePubMedPMC
- 47. Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38: 787–793. PMID: 16804544. ArticlePubMed
- 48. Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn 2008; 10: 13–27. PMID: 18165277. ArticlePubMedPMC
- 49. Kim JH, Shin SH, Kwon HJ, Cho NY, Kang GH. Prognostic implications of CpG island hypermethylator phenotype in colorectal cancers. Virchows Arch 2009; 455: 485–494. PMID: 19911194. ArticlePubMed
- 50. Toyota M, Ahuja N, Suzuki H, et al. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res 1999; 59: 5438–5442. PMID: 10554013. PubMed
- 51. Enomoto S, Maekita T, Tsukamoto T, et al. Lack of association between CpG island methylator phenotype in human gastric cancers and methylation in their background non-cancerous gastric mucosae. Cancer Sci 2007; 98: 1853–1861. PMID: 17900260. ArticlePubMed
- 52. Chang MS, Uozaki H, Chong JM, et al. CpG island methylation status in gastric carcinoma with and without infection of Epstein-Barr virus. Clin Cancer Res 2006; 12: 2995–3002. PMID: 16707594. ArticlePubMed
- 53. Kang GH, Lee S, Kim WH, et al. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol 2002; 160: 787–794. PMID: 11891177. ArticlePubMedPMC
- 54. Kusano M, Toyota M, Suzuki H, et al. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein-Barr virus. Cancer 2006; 106: 1467–1479. PMID: 16518809. ArticlePubMed
- 55. Oue N, Oshimo Y, Nakayama H, et al. DNA methylation of multiple genes in gastric carcinoma: association with histological type and CpG island methylator phenotype. Cancer Sci 2003; 94: 901–905. PMID: 14556664. ArticlePubMed
- 56. An C, Choi IS, Yao JC, et al. Prognostic significance of CpG island methylator phenotype and microsatellite instability in gastric carcinoma. Clin Cancer Res 2005; 11(2 Pt 1):656–663. PMID: 15701853. ArticlePubMedPDF
- 57. Park SY, Kook MC, Kim YW, et al. CpG island hypermethylator phenotype in gastric carcinoma and its clinicopathological features. Virchows Arch 2010; 457: 415–422. PMID: 20737169. ArticlePubMed
- 58. Imai S, Koizumi S, Sugiura M, et al. Gastric carcinoma: monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proc Natl Acad Sci U S A 1994; 91: 9131–9135. PMID: 8090780. ArticlePubMedPMC
- 59. Fukayama M, Hayashi Y, Iwasaki Y, et al. Epstein-Barr virus-associated gastric carcinoma and Epstein-Barr virus infection of the stomach. Lab Invest 1994; 71: 73–81. PMID: 8041121. PubMed
- 60. Fukayama M, Hino R, Uozaki H. Epstein-Barr virus and gastric carcinoma: virus-host interactions leading to carcinoma. Cancer Sci 2008; 99: 1726–1733. PMID: 18616681. ArticlePubMed
- 61. Kang GH, Lee S, Cho NY, et al. DNA methylation profiles of gastric carcinoma characterized by quantitative DNA methylation analysis. Lab Invest 2008; 88: 161–170. PMID: 18158559. ArticlePubMed
- 62. Etoh T, Kanai Y, Ushijima S, et al. Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am J Pathol 2004; 164: 689–699. PMID: 14742272. ArticlePubMedPMC
- 63. Hino R, Uozaki H, Murakami N, et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res 2009; 69: 2766–2774. PMID: 19339266. ArticlePubMed
- 64. Kim H, An JY, Noh SH, Shin SK, Lee YC, Kim H. High microsatellite instability predicts good prognosis in intestinal-type gastric cancers. J Gastroenterol Hepatol 2011; 26: 585–592. PMID: 21332554. ArticlePubMed
- 65. Gylling A, Abdel-Rahman WM, Juhola M, et al. Is gastric cancer part of the tumour spectrum of hereditary non-polyposis colorectal cancer? A molecular genetic study. Gut 2007; 56: 926–933. PMID: 17267619. ArticlePubMedPMC
- 66. Kang GH, Lee S, Kim JS, Jung HY. Profile of aberrant CpG island methylation along the multistep pathway of gastric carcinogenesis. Lab Invest 2003; 83: 635–641. PMID: 12746473. ArticlePubMed
- 67. Lee JH, Park SJ, Abraham SC, et al. Frequent CpG island methylation in precursor lesions and early gastric adenocarcinomas. Oncogene 2004; 23: 4646–4654. PMID: 15064707. ArticlePubMed
- 68. Ling ZQ, Tanaka A, Li P, et al. Microsatellite instability with promoter methylation and silencing of hMLH1 can regionally occur during progression of gastric carcinoma. Cancer Lett 2010; 297: 244–251. PMID: 20831982. ArticlePubMed
- 69. Saito Y, Liang G, Egger G, et al. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006; 9: 435–443. PMID: 16766263. ArticlePubMed
- 70. Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J 2011; 278: 1598–1609. PMID: 21395977. ArticlePubMed
- 71. Saito Y, Suzuki H, Tsugawa H, et al. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene 2009; 28: 2738–2744. PMID: 19503096. ArticlePubMed
- 72. Tsukamoto Y, Nakada C, Noguchi T, et al. MicroRNA-375 is downregulated in gastric carcinomas and regulates cell survival by targeting PDK1 and 14-3-3zeta. Cancer Res 2010; 70: 2339–2349. PMID: 20215506. ArticlePubMed
- 73. Wada R, Akiyama Y, Hashimoto Y, Fukamachi H, Yuasa Y. miR-212 is downregulated and suppresses methyl-CpG-binding protein MeCP2 in human gastric cancer. Int J Cancer 2010; 127: 1106–1114. PMID: 20020497. ArticlePubMed
- 74. Tsai KW, Hu LY, Wu CW, et al. Epigenetic regulation of miR-196b expression in gastric cancer. Genes Chromosomes Cancer 2010; 49: 969–980. PMID: 20662076. ArticlePubMed
- 75. Chen Q, Chen X, Zhang M, Fan Q, Luo S, Cao X. miR-137 is frequently down-regulated in gastric cancer and is a negative regulator of Cdc42. Dig Dis Sci 2011; 56: 2009–2016. PMID: 21221794. ArticlePubMed
- 76. Shen R, Pan S, Qi S, Lin X, Cheng S. Epigenetic repression of microRNA-129-2 leads to overexpression of SOX4 in gastric cancer. Biochem Biophys Res Commun 2010; 394: 1047–1052. PMID: 20331975. ArticlePubMed
- 77. Ando T, Yoshida T, Enomoto S, et al. DNA methylation of microRNA genes in gastric mucosae of gastric cancer patients: its possible involvement in the formation of epigenetic field defect. Int J Cancer 2009; 124: 2367–2374. PMID: 19165869. ArticlePubMed
- 78. Suzuki H, Yamamoto E, Nojima M, et al. Methylation-associated silencing of microRNA-34b/c in gastric cancer and its involvement in an epigenetic field defect. Carcinogenesis 2010; 31: 2066–2073. PMID: 20924086. ArticlePubMed
References
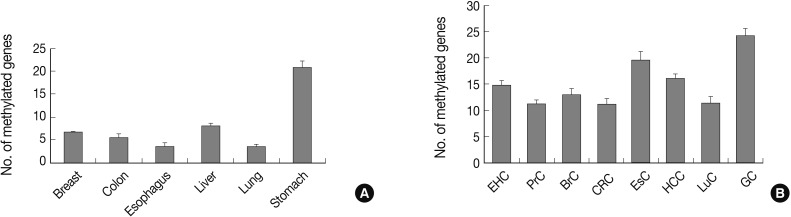
Fig. 1Bar graphs display the number of methylated genes in cancer-associated normal tissue (A) and cancer tissue (B). The error bars indicate the standard error of the mean. Forty-one genes are analyzed for their methylation status in tissue samples from eight human cancer tissue types and six types of cancer-associated normal tissue using the MethyLight assay. EHC, extrahepatic bile duct cancer; PrC, prostate adenocarcinoma; BrC, breast adenocarcinoma; CRC, colorectal adenocarcinoma; EsC, esophageal adenocarcinoma; HCC, hepatocellular carcinoma; LuC, lung adenocarcinoma; GC, gastric adenocarcinoma.
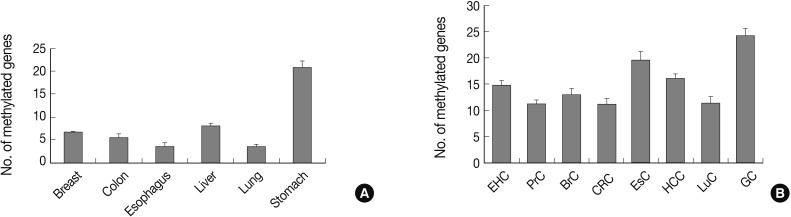
Figure & Data
References
Citations
Citations to this article as recorded by 
- Genome-wide characterization of extrachromosomal circular DNA in gastric cancer and its potential role in carcinogenesis and cancer progression
Xianming Jiang, Xiaoguang Pan, Wenchao Li, Peng Han, Jiaying Yu, Jing Li, Haoran Zhang, Wei Lv, Ying Zhang, Yulong He, Xi Xiang
Cellular and Molecular Life Sciences.2023;[Epub] CrossRef - Analysis of DNA methylation in endometrial biopsies to predict risk of endometrial cancer
Francesco Multinu, Jun Chen, Joseph D. Madison, Michelle Torres, Jvan Casarin, Daniel Visscher, Viji Shridhar, Jamie Bakkum-Gamez, Mark Sherman, Nicolas Wentzensen, Andrea Mariani, Marina Walther-Antonio
Gynecologic Oncology.2020; 156(3): 682. CrossRef - Helicobacter pylori severely reduces expression of DNA repair proteins PMS2 and ERCC1 in gastritis and gastric cancer
Yasir Raza, Ayaz Ahmed, Adnan Khan, Arif Ali Chishti, Syed Shakeel Akhter, Muhammad Mubarak, Carol Bernstein, Beryl Zaitlin, Shahana Urooj Kazmi
DNA Repair.2020; 89: 102836. CrossRef - Genomic and Epigenomic Profiling of High-Risk Intestinal Metaplasia Reveals Molecular Determinants of Progression to Gastric Cancer
Kie Kyon Huang, Kalpana Ramnarayanan, Feng Zhu, Supriya Srivastava, Chang Xu, Angie Lay Keng Tan, Minghui Lee, Suting Tay, Kakoli Das, Manjie Xing, Aliya Fatehullah, Syed Muhammad Fahmy Alkaff, Tony Kiat Hon Lim, Jonathan Lee, Khek Yu Ho, Steven George Ro
Cancer Cell.2018; 33(1): 137. CrossRef - Decreased Methylation of IFNAR Gene Promoter from Peripheral Blood Mononuclear Cells Is Associated with Oxidative Stress in Chronic Hepatitis B
Jing-wen Wang, Jing-wei Wang, Jun Zhang, Chen-si Wu, Yu Fang, Wei-wei Su, Yu-chen Fan, Kai Wang
Journal of Interferon & Cytokine Research.2018; 38(11): 480. CrossRef - Genomic landscape of gastric cancer: molecular classification and potential targets
Jiawei Guo, Weiwei Yu, Hui Su, Xiufeng Pang
Science China Life Sciences.2017; 60(2): 126. CrossRef - Hypermethylation of the galectin-3 promoter is associated with poor prognosis of acute-on-chronic hepatitis B liver failure
Jing Zhao, Yu-Chen Fan, Xin-Yuan Liu, Ze-Hua Zhao, Feng Li, Kai Wang
Digestive and Liver Disease.2017; 49(6): 664. CrossRef - Proteomics-Based Identification and Analysis of Proteins Associated with Helicobacter pylori in Gastric Cancer
Jianjiang Zhou, Wenling Wang, Yuan Xie, Yan Zhao, Xian Chen, Wenjie Xu, Yan Wang, Zhizhong Guan, Hiromu Suzuki
PLOS ONE.2016; 11(1): e0146521. CrossRef - Lack of Correlation between Aberrant p16, RAR-β2, TIMP3, ERCC1, and BRCA1 Protein Expression and Promoter Methylation in Squamous Cell Carcinoma Accompanying Candida albicans-Induced Inflammation
Yui Terayama, Tetsuro Matsuura, Kiyokazu Ozaki, Javier S Castresana
PLOS ONE.2016; 11(7): e0159090. CrossRef - Helicobacter pylori CagA induces tumor suppressor gene hypermethylation by upregulating DNMT1 via AKT-NFκB pathway in gastric cancer development
Bao-gui Zhang, Lei Hu, Ming-de Zang, He-xiao Wang, Wei Zhao, Jian-fang Li, Li-ping Su, Zhifeng Shao, Xiaodong Zhao, Zheng-gang Zhu, Min Yan, Bingya Liu
Oncotarget.2016; 7(9): 9788. CrossRef - Promoter methylation status and expression of PPAR-γ gene are associated with prognosis of acute-on-chronic hepatitis B liver failure
Ze-Hua Zhao, Yu-Chen Fan, Qi Zhao, Cheng-Yun Dou, Xiang-Fen Ji, Jing Zhao, Shuai Gao, Xin-You Li, Kai Wang
Clinical Epigenetics.2015;[Epub] CrossRef - Role of epigenetics in EBV regulation and pathogenesis
Hans Helmut Niller, Zsófia Tarnai, Gábor Decsi, Ádám Zsedényi, Ferenc Bánáti, Janos Minarovits
Future Microbiology.2014; 9(6): 747. CrossRef - Helicobacter pylori Induces Hypermethylation of CpG Islands Through Upregulation of DNA Methyltransferase: Possible Involvement of Reactive Oxygen/Nitrogen Species
Hye-Kyung Na, Jeong-Hwa Woo
Journal of Cancer Prevention.2014; 19(4): 259. CrossRef - Mallory–Denk Body (MDB) formation modulates ufmylation expression epigenetically in alcoholic hepatitis (AH) and non-alcoholic steatohepatitis (NASH)
Hui Liu, Ming Gong, Barbara A. French, Jun Li, Brittany Tillman, Samuel W. French
Experimental and Molecular Pathology.2014; 97(3): 477. CrossRef - RETRACTED ARTICLE: Role of p16 gene promoter methylation in gastric carcinogenesis: a meta-analysis
He-Ling Wang, Ping-Yi Zhou, Peng Liu, Yu Zhang
Molecular Biology Reports.2014; 41(7): 4481. CrossRef - Exportin 4 gene expression and DNA promoter methylation status in chronic hepatitis B virus infection
F. Zhang, Y.‐C. Fan, N.‐N Mu, J. Zhao, F.‐K. Sun, Z.‐H. Zhao, S. Gao, K. Wang
Journal of Viral Hepatitis.2014; 21(4): 241. CrossRef - Microarray-based DNA methylation study of Ewing’s sarcoma of the bone
HYE-RIM PARK, WOON-WON JUNG, HYUN-SOOK KIM, YONG-KOO PARK
Oncology Letters.2014; 8(4): 1613. CrossRef - Pathologic Diagnosis of Gastric Intestinal Metaplasia
Nari Shin, Do Youn Park
The Korean Journal of Helicobacter and Upper Gastrointestinal Research.2013; 13(2): 84. CrossRef
 PubReader
PubReader Cite this Article
Cite this Article