Molecular Dimensions of Gastric Cancer: Translational and Clinical Perspectives
Article information
Abstract
Gastric cancer is a global health burden and has the highest incidence in East Asia. This disease is complex in nature because it arises from multiple interactions of genetic, local environmental, and host factors, resulting in biological heterogeneity. This genetic intricacy converges on molecular characteristics reflecting the pathophysiology, tumor biology, and clinical outcome. Therefore, understanding the molecular characteristics at a genomic level is pivotal to improving the clinical care of patients with gastric cancer. A recent landmark study, The Cancer Genome Atlas (TCGA) project, showed the molecular landscape of gastric cancer through a comprehensive molecular evaluation of 295 primary gastric cancers. The proposed molecular classification divided gastric cancer into four subtypes: Epstein-Barr virus–positive, microsatellite unstable, genomic stable, and chromosomal instability. This information will be taken into account in future clinical trials and will be translated into clinical therapeutic decisions. To fully realize the clinical benefit, many challenges must be overcome. Rapid growth of high-throughput biology and functional validation of molecular targets will further deepen our knowledge of molecular dimensions of this cancer, allowing for personalized precision medicine.
Gastric cancer (GC) is one of the most common and fatal diseases, and nearly two-thirds of the cases are concentrated in East Asia [1]. In Korea, GC is the second most common malignancy after thyroid cancer. It was ranked as the third cause of cancer mortality in 2012 [2]. An estimate of 35,000 people are expected to be newly diagnosed, and about 7,500 patients will die in 2015 [3]. According to the National Cancer Screening Program that began in 1999, the proportion of early gastric cancer (EGC) has increased, and the prognosis of EGC is favorable even without additional treatment after surgery [4,5]. Radical D2 surgery with adjuvant chemotherapy has been established as a standard treatment for locally advanced gastric cancer (AGC), and has improved the prognosis of AGC [6-8]. However, about half of patients with AGC experience recurrence, and the proportion of patients who benefit from adjuvant chemotherapy is around 20% [9-14], with cases of metastasis being the least amenable to treatment [15,16].
GC is a heterogeneous malignancy, which is the main reason for its different prognoses in patients with same clinical stage and for its diverse responses to the standard treatment. It is now widely appreciated that genomic complexity and heterogeneity are fundamental causes of tumor phenotypic characteristics determining clinical outcomes. Therefore, understanding of the molecular and genetic characteristics is essential in effective and personalized management of GC.
GC has been classified according to histo-morphologic features. The World Health Organization (WHO) classifies GC into papillary, tubular, mucinous, and poorly cohesive carcinomas [17], while the Lauren classification divides GC into intestinal, diffuse, and mixed types [18]. However, these classification systems do not satisfactorily provide information relevant to clinical utilities and treatment guidelines. Over the past decades, the molecular landscape of GC has been shaped, and this review focuses on the molecular features of GC that can be translated into clinical use in order to guide precise therapeutic decisions. Details of surgical, adjuvant or peri-operative chemotherapy, and radiotherapy are beyond the scope of this review and readers would be directed to other reviews dealing with these issues [19]. The recent The Cancer Genome Atlas (TCGA) study [20], a landmark study dividing GC into four subtypes based on multi-dimensional profiling, (1) Epstein-Barr virus (EBV) tumor, (2) microsatellite unstable (MSI) tumor, (3) genomically stable (GS) tumor, and (4) chromosomal instability (CIN), is used as a roadmap for this review. We also provide the current status of recent and ongoing clinical trials that focus on genomic alterations in molecular subtypes for targeted therapeutics (summarized in Table 1) [15,16,21-34].
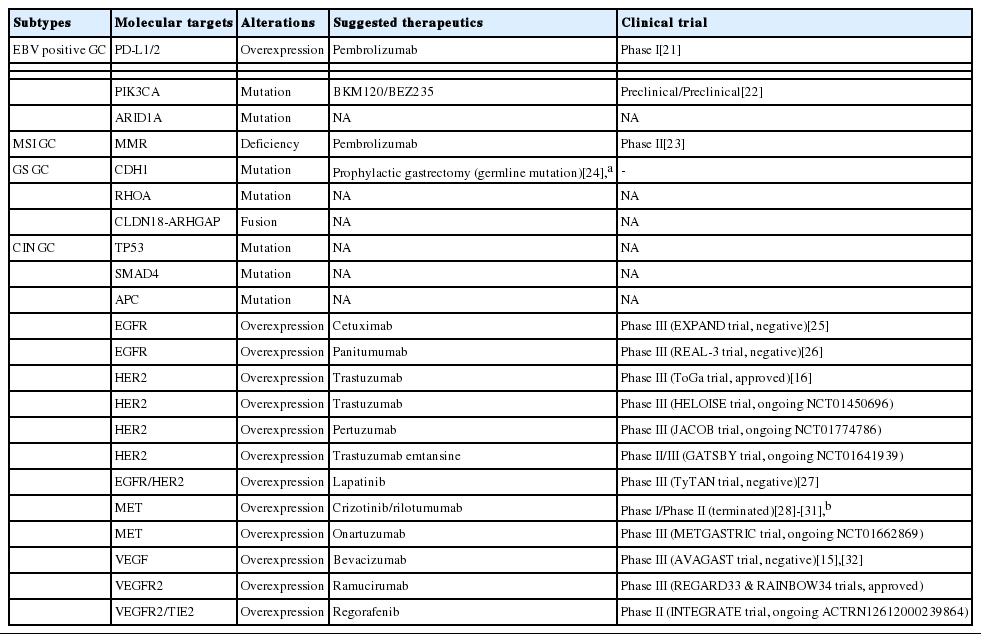
Current status of targeted therapies based on molecular alterations according to GC subtype
EPSTEIN-BARR VIRUS–POSITIVE GASTRIC CANCER
The incidence of EBV-positive GC has been reported to be around 10% [20,35] and harbors a higher prevalence of DNA hypermethylation than other subtypes [20,36]. The reason for extraordinary DNA hypermethylation seems to be a cellular reaction to the viral infection [37]. EBV-positive GC also has strong signatures of interleukin 12-mediated signaling events, which reflects high immune cell infiltration [20,38]. Intriguingly, tumors of this subtype exhibit CD274 and PDCD1LG2 amplification of which proteins PD-L1 and PD-L2, respectively, are related to immune suppressive functions, particularly immune checkpoints. Further, amplification at the 9p24.1 locus containing JAK2, which encodes an oncogenic receptor tyrosine kinase (RTK), is also a potential therapeutic target. Those findings seemed to support the rationale that this type of GC would be a relevant target of RTK and immune checkpoints inhibitors [20,37]. A recent phase I study showed that pembrolizumab (MK-3475), one of the anti–PD-1/2 immune checkpoint inhibitors, provided antitumor activity in patients with AGC that expressed PD-L1 [21]. Predilection of EBV-positive GC for PIK3CA mutation, which is related to the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway, has also been reported [20]. Frequent PIK3CA mutations warrant evaluation of PI3K inhibitors in EBV-positive GC. Although still in preclinical stages, BKM 120, which is a direct PIK3CA inhibitor, and BEZ235, a dual PIK3CA and mammalian target of rapamycin (mTOR) inhibitor, have been reported to reduce cell viability and induce apoptosis in GC cell lines [22]. Additionally, the ARID1A mutation was detected in 10% of GC39 and was found most frequently in EBV-positive GC [20]. This mutation encodes a component of SWI/SNF complex and acts as a tumor suppressor in cancer [39]. Recently, it was reported that EZH2 inhibitor could be a novel therapeutic targeting ARID1A-mutated cancers [40]. Therefore, the ARID1A mutation provides another clinically actionable genetic alteration in EBV-positive GC that should be validated in a clinical study. Based on these findings, the molecular characteristics of EBV-positive GC are distinct from those of other GC subtypes, and some of the genetic alterations can be therapeutically exploited.
MICROSATELLITE UNSTABLE GASTRIC CANCER
MSI GC is related to the loss of function of mismatch repair (MMR) genes and is associated with older age, female gender, intestinal type, and less aggressive tumor stages [20,41,42]. Because the function of the MMR mechanism is defective mainly due to MLH1 silencing by promoter hypermethylation [20], this subtype has more mutations per megabase (Mb) compared to other types of GC. Intriguingly, MSI tumors possess common alterations in major histocompatibility complex (MHC) class I-related genes, including HLA-B and B2M. Since these MHC class I genes function in proper antigen presentation to the host immune system, these genomic alterations could provide hypermutated MSI GC with the selective advantage of immune surveillance evasion.
The incidence of MSI GC was previously reported to be 8.5%–37.8% [43]. While the prognosis of MSI GC was not assessed in comparison with those of other molecular subtypes in a TCGA study [20], a meta-analysis [43] and a recent study regarding the molecular classification of GC reported that MSI GC had the best overall prognosis with the lowest recurrence rate [44]. Importantly, the prognosis of MSI GC was prominent in the population treated with surgery alone. Indeed, the prognosis of MSI GC without chemotherapy was similar to that of patients who received chemotherapy after surgery [45], implying that the MSI subtype is unresponsive to chemotherapy in an adjuvant setting [41], like MSI colon cancer [46,47]. Additionally, a recent phase II study that evaluated the clinical utility of pembrolizumab showed that MMR status predicted the benefit of pembrolizumab [23], and a higher mutational load was reported to be related to positive response to anti-CTLA-4 in melanoma [48] and PD-1 antibody in non-small cell lung cancer [49]. The legitimate explanation might be that immune infiltrate related with mutation was directed at neoantigens, and recognition plays an important role in the antitumor immune response [23]. Consequently, MSI status is a promising biomarker to predict the prognosis and responses to immune checkpoint inhibitor as well as chemotherapy in GC, as in colon cancer [50]. There is no consensus on the definition of GC-specific MSI in clinical settings at this time, and studies have used different criteria to define MSI[20,43,45,51,52]. Thus, it is necessary to establish appropriate analysis standards for MSI status in GC for precise detection and translation of “MSI-ness” for clinical therapeutic decisions.
GENOMICALLY STABLE GASTRIC CANCER
The GS subtype of GC is best represented as a diffuse type of GC, with lower mutation burden compared to other subtypes and occurring at a relatively early age [20]. CDH1 mutation is one of the representative mutations in the GS subtype. CDH1 germline mutations are known to be related to hereditary diffuse GC. When patients harbor pathogenic hotspot mutations in CDH1, prophylactic gastrectomy is recommended [24]. However, only two CDH1 mutations, neither of which is a pathogenic hotspot mutation, were identified in a recent TCGA study [20]. Another study reported that somatic alterations of CDH1 were present in approximately 30% of GC cases, and structural alterations in CDH1 were related to poor prognosis [53]. In addition to CDH1 mutations, GS subtype tumors have RHOA mutations and CLDN18-ARHGAP 6 or 26 fusions [20,54,55]. RHOA is known to modulate downstream Rho signaling, and its mutation imparts resistance to anoikis, a form of programmed cell death [54]. Also, RHOA acts to control actin-myosin-dependent cell contractility and motility [56,57]; thus, its mutation might contribute to dispersed growth and poorly cohesive patterns of diffuse type GC [20], which is associated with poor prognosis. Thus, the RHOA mutation could be a good candidate for new approaches targeting GS subtype GC [37]. CLDN18-ARHGAP6 or 26 fusions are mutually exclusive to RHOA and CDH1 mutation among GS tumors. The discovery of recurrent interchromosomal translocation between CLDN18 and ARHGAP26 further implies biological significance of cell adhesion and deregulated Rho signaling in GS tumors since CLDN18 is involved in intercellular tight junction structure, and ARHGAP26, a GTPase-activating protein, imparts Rho signaling activation by facilitating the conversion of Rho GTPases to the GDP state. A recent study reported that this type of fusion in epithelial cells mediates epithelial disintegration and is related to epithelial-mesenchymal transition (EMT) [58]. Therefore, the novel discoveries of RHOA mutation and CLDN18-ARHGAP26 fusion could be exploited to develop new therapeutic strategies against GS subtype tumors [37]. which are known to harbor the poorest prognosis of all GC tumors [44]. However, translating those new strategies to clinical practice is in the early stages and is largely lacking evidence of functional validity. Additionally, there have been no clinical trials to assess the efficacy of targeting those genomic alterations in GS subtype tumors.
CHROMOSOMAL INSTABILITY GASTRIC CANCER
CIN subtype GC is related to intestinal type histology, frequent TP53 mutations, and amplification of RTKs [20]. TP53 mutation is the most frequently detected mutation in GC, occurring in up to 50% of all cases [59] and 71% of cases of CIN subtype GC [20]. TP53 mutation is associated with high levels of somatic copy number variations in both chromosomal and focal gene regions [60]. Also, other canonical tumor suppressor genes such as SMAD4 and APC have been reported to be mutated in GC [61]. Since tumor suppressor genes are regarded as poor candidates for targeted therapy development, alterations in RTKs will be discussed in this review.
EPIDERMAL GROWTH FACTOR RECEPTOR
The human epidermal growth factor receptors (HER) are a family of four transmembrane RTKs, ErbB1 (epidermal growth factor receptor, EGFR), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4) [62], that regulate diverse downstream signaling pathways and play an important role in GC development and progression. A biomarker study from Adjuvant Chemotherapy Trial of S-1 for Gastric Cancer (ACTS-GC) [14], a phase III randomized controlled trial (RCT) that compared the effect of adjuvant S-1 over surgery alone in locally AGC, showed that EGFR overexpression was related to poor prognosis but was not found to be the case for HER2 [63]. There have been two RCTs that investigated the benefit of EGFR inhibition (EXPAND [25] trial for cetuximab and REAL-3 trial [26] for panitumumab as first-line therapy); however, both trials failed to prove the additional clinical benefit of anti-EGFR antibody over standard chemotherapy.
HER2
The success of a clinical trial that investigated the effects of trastuzumab targeting HER2 in HER2-overexpressed GC patients resulted in changes in clinical practice. Addition of trastuzumab to chemotherapy as the first-line treatment of metastatic GC improved overall survival [16]. However, an updated survival analysis showed that the benefit of the trastuzumab decreased over time, the difference in median overall survival was reduced from 2.7 to 1.4 months, and the hazard ratio increased from 0.74 in primary analysis to 0.80 [64]. This raises a concern, requiring further investigation to clarify the clinical benefit of trastuzumab in HER2-overexpressing GC patients. Indeed, two first-line therapy trials are underway to investigate the effect of addition of pertuzumab to a standard HER2 targeting regimen and the effect of two dose levels of trastuzumab (JACOB and HELIOSE, respectively).
The frequency of HER2 mutation was reported to be 5% (9/180) in GC, and the relationship between HER2 mutation and responsiveness to trastuzumab has not yet been determined. Another clinically important issue regarding HER2 mutation and amplification might be derived from a recent EGFR biomarker study in non-small cell lung cancer patients receiving gefitinib, where EGFR mutation and amplification correlated with prolonged progression-free survival [65]. Based on this, it might be worthwhile to investigate the clinical benefit of HER2 inhibitor in HER2-mutated and amplified GC as alternative candidates for HER2-targeted therapy [66]. A phase III trial (TYTAN) was conducted to investigate the benefit of lapatinib, a dual inhibitor of EGFR and HER2, as a second-line therapy for AGC. Although overall survival was not significantly different, post hoc analysis demonstrated that the HER2 immunohistochemistry 3+ subgroup showed statistically significant prolongation of overall survvial [27].
KRAS
KRAS is one of the members of the RAS family, and its mutation plays an important role in tumorigenesis by activating downstream pathways such as PI3K and RAF. The frequency of KRAS mutations in GC was reported as 1.5%–5.8%, and most of them were transversions [67]. Overexpression of wild-type KRAS seemed to be related to acquired resistance to inhibitors of other tyrosine kinase in GC cells [68]. A phase II trial that evaluated the efficacy of selumetinib, an inhibitor of MEK1/MEK2, downstream of KRAS, for KRAS-mutant non-small cell lung cancer demonstrated promising efficacy and thereby warrants further clinical investigation [69]. Thus, a MEK inhibitor could be a potential therapeutic agent for targeting KRAS-mutated GC; however, evidence from a clinical trial is required.
MESENCHYMAL EPITHELIAL TRANSITION FACTOR
Mesenchymal epithelial transition factor (MET) amplification was not common (2%, 10/489) in GC it was reported to be related with poor prognosis [28]. However, two studies have reported the possibility of targeted therapy for MET-positive GC. An expanded phase I cohort study showed that patients with MET amplification had a favorable response to crizotinib (PF-02341066), a MET/anaplastic lymphoma kinase tyrosine kinase inhibitor [28]. Furthermore, rilotumumab (AMG 102), a fully humanized monoclonal antibody against hepatocyte growth factor/MET, demonstrated favorable overall survival especially for patients with MET-positive GC [29]. Based on those results, subsequent trials were conducted (RILOMET-1 [30] and NCT02137343); however, all Amgen-sponsored clinical trials of rilotumumab in AGC were terminated based on a pre-planned safety review by the data monitoring committee due to an increase in death with the study drug [31]. Currently, a small-molecule MET inhibitor is under investigation for MET-amplified GC.
BRAF
BRAF mutations are related to tumorigenesis, and dysregulated BRAF activity instigates abnormal cell growth and proliferation through MEK and ERK pathways [70]. The specific mutation BRAFV600E is the most common type of BRAF mutation in melanoma, and vemurafenib, a BRAF inhibitor, was found to be beneficial in patients with BRAFV600E-mutated melanoma [71]. BRAF mutations are rarely observed in GC, with only 2.2% (7/319) of patients demonstrating BRAF mutation, most (five of seven BRAF mutations) of which were BRAFV599M [72]; in addition, there were no BRAF mutations among 167 patients in the REAL-3 trial [26]. Furthermore, only 0.2% of patients (1/508) with GC had a BRAFV600E mutation in another study [67]. Therefore, it is not yet clear if BRAF mutation is a driver mutation in GC.
VASCULAR ENDOTHELIAL GROWTH FACTOR RECEPTOR
Vascular endothelial growth factor receptor (VEGFR) expression is closely related to angiogenesis in tumorigenesis. Since angiogenesis is critical for tumor growth and metastasis, it is a therapeutic target for many cancer types. Bevacizumab, a monoclonal antibody that inhibits VEGF (VEGF-A), showed survival benefits in advanced colorectal cancer [73] and non-small cell lung cancer [74]. Also, a phase II trial that showed efficacy of bevacizumab for advanced gastro-esophageal cancer seemed to reinforce the success of targeted therapy against the VEGF pathway in gastric cancer [75]. Despite initial enthusiasm regarding its use, bevacizumab combined with chemotherapy as a first-line therapy did not improve the overall survival of patients with GC in the AVAGAST trial. However, the more recent RAINBOW [34] and REGARD [33] trials, which evaluated benefits of ramucirumab (antibody targeting VEGFR2) as second-line and first-line therapies, respectively, reported improved overall survival in GC patients. Intriguingly, subgroup analyses of the trials showed that benefit from VEGF-targeted therapy was observed mainly in non-Asian patients. Also, a subsequent biomarker study of AVAGAST showed that plasma VEGF-A and neuropilin-1 levels could be prognostic and predictive of bevacizumab treatment in a non-Asian population [32]. Those findings imply that GC is a complex and heterogeneous disease across the globe, which affects the response to anti-angiogenesis treatment and potentially other target therapies [37,76].
CANCER STEM CELL-RELATED PATHWAYS
Cancer stem cells that initiate tumorigenesis through self-renewal and differentiation are emerging concepts in cancer research. Such cells activate EMT, oncogenic pathways, and embryogenic pathways [77,78]. Also, these cells are known to be resistant to chemotherapy and radiotherapy, while Wnt, Notch, and Hedgehog pathways are crucial to the maintenance of cancer stem cells. Transcriptional factors such as Snail, Slug, Twist, and Zeb1/2 coordinate the EMT, while transforming growth factor β (TGF-β) is a central signaling pathway related to transformation into EMT [79,80]. TGF-β can act as a proto-oncogene, driving matrix deposit, stimulating EMT and stem cell renewal, and inhibiting apoptosis through transactivation of EGFR [81,82]. Also, its downstream signaling pathway, PI3K/Akt/mTOR, aids in cancer stem cell maintenance [83]. Thus, targeting this pathway with appropriate agents such as metformin would inhibit cellular transformation and selectively kill cancer stem cells, as previously demonstrated in breast cancer [84]. Also, a recent report showed that patients with GC treated with metformin for diabetes mellitus had better survival compared to those treated with diabetic medications other than metformin [85]. GC stem cells express CD133, CD44, aldehyde dehydrogenase 1 (ALDH1), and ATP-binding cassette sub-family G member 2 (ABCG2). CD44 and ALDH1 have been reported to be related to resistance to chemotherapy and radiotherapy [86]. Amplification of the gene that encodes the GC stem cell marker CD44 was observed in a TCGA study [20], suggesting the potential of exploiting genomic alteration in future development of cancer stem cell-directed therapies.
CONCLUSIONS AND FUTURE PERSPECTIVES
Through extraordinary efforts over the past decades, our knowledge on GC has advanced considerably, and standardized multidisciplinary treatment has improved the prognosis of GC. However, clinical development of targeted therapy in GC remains inferior to those of other cancer types such as lung, breast, and colon cancer in terms of genetic sequencing and molecular therapeutics [87]. Most of the targeted therapies have been investigated without patient selection based on a biomarker, and the results have been disappointing, with only a few targeted agents [16,33,34] showing benefit to patient survival at one year even after treatment for metastasis or recurrent GC. In the upcoming years, accumulation of genomic information and knowledge about molecular pathogenesis of GC will be accelerated through high-throughput systems biology, and the treatment will be focused on targeting specific GC subtypes based on specific molecular characteristics (e.g., somatic driver alterations and amplification). Presently, one of the hurdles in this achievement is the integration of knowledge from various disciplines and its translation into daily clinical practice. To achieve a sensible reduction in mortality due to this deadly disease, transdisciplinary cooperation among clinicians, pathologists, bioiformaticians, computational biologists, and genomicists is required.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI3C2162), and by the National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (1020390, 1320360).