Pathogenesis of Focal Segmental Glomerulosclerosis
Article information
Abstract
Focal segmental glomerulosclerosis (FSGS) is characterized by focal and segmental obliteration of glomerular capillary tufts with increased matrix. FSGS is classified as collapsing, tip, cellular, perihilar and not otherwise specified variants according to the location and character of the sclerotic lesion. Primary or idiopathic FSGS is considered to be related to podocyte injury, and the pathogenesis of podocyte injury has been actively investigated. Several circulating factors affecting podocyte permeability barrier have been proposed, but not proven to cause FSGS. FSGS may also be caused by genetic alterations. These genes are mainly those regulating slit diaphragm structure, actin cytoskeleton of podocytes, and foot process structure. The mode of inheritance and age of onset are different according to the gene involved. Recently, the role of parietal epithelial cells (PECs) has been highlighted. Podocytes and PECs have common mesenchymal progenitors, therefore, PECs could be a source of podocyte repopulation after podocyte injury. Activated PECs migrate along adhesion to the glomerular tuft and may also contribute to the progression of sclerosis. Markers of activated PECs, including CD44, could be used to distinguish FSGS from minimal change disease. The pathogenesis of FSGS is very complex; however, understanding basic mechanisms of podocyte injury is important not only for basic research, but also for daily diagnostic pathology practice.
Focal segmental glomerulosclerosis (FSGS) typically presents with nephrotic range proteinuria and, as its name implies, shows obliteration or collapse of glomerular capillary loops by increased extracellular matrix in some glomeruli only, and the capillary injury does not occupy the entire glomerulus involved. In primary FSGS, direct podocyte injury is postulated to result in sclerosis. Though morphologically similar, secondary FSGS develops due to varying injuries, conditions such as obesity, renal mass reduction, drug toxicity, viral infection, familial genetic background, hypertension-related injury, chronic pyelonephritis, or healing of pauciimmune necrotizing crescentic injury.
FSGS has heterogeneous morphology (Fig. 1). Several attempts have been made to classify FSGS according to the pattern and intraglomerular distribution of sclerotic lesions. The Columbia Classification proposed in 2004 divides FSGS into collapsing, tip, cellular, perihilar, and not otherwise specified (NOS) variants [1]. In collapsing variant of FSGS, glomerular capillary collapse accompanied by podocyte hypertrophy and hyperplasia is observed in at least one glomerulus. The presence of other variants in the remaining glomeruli does not alter the diagnosis. This variant has been characterized by its aggressive behavior [2]. While collapsing FSGS is a histologic feature of human immunodeficiency virus–associated nephropathy (HIVAN), it has also been related to various conditions such as other viral infection (parvovirus B19 [3]) and drugs (pamidronate [4], interferons [5], and anthracyclin [6]), or it can be idiopathic [7]. The tip variant means that the segmental lesion involves the outer 25% of the glomerular tuft next to the tubular pole, i.e., tip portion, in the absence of collapsing or perihilar lesions [1]. Tip variant has a favorable prognosis and good response to therapy in most series [8]. However, contrary reports have also been published [9]. In the cellular variant, at least one glomerulus shows segmental endocapillary hypercellularity, but not in a tip location [1]. The perihilar variant is diagnosed when more than half of the sclerotic glomeruli have sclerosis or hyalinosis in the perihilar area [1]. In a study on Korean adults, the incidence of each subtype was 63.1%, 18.0%, 15.3%, 2.7%, and 0.9% for NOS, tip, perihilar, cellular and collapsing variant, respectively [10]. In Korean children, the incidence was 72.7%, 6.1%, 9.1%, 1.5%, and 10.6% for each subtype [11]. The low incidence of collapsing lesions in adults may partly be associated with the low frequency of HIVAN in Asia [12,13].
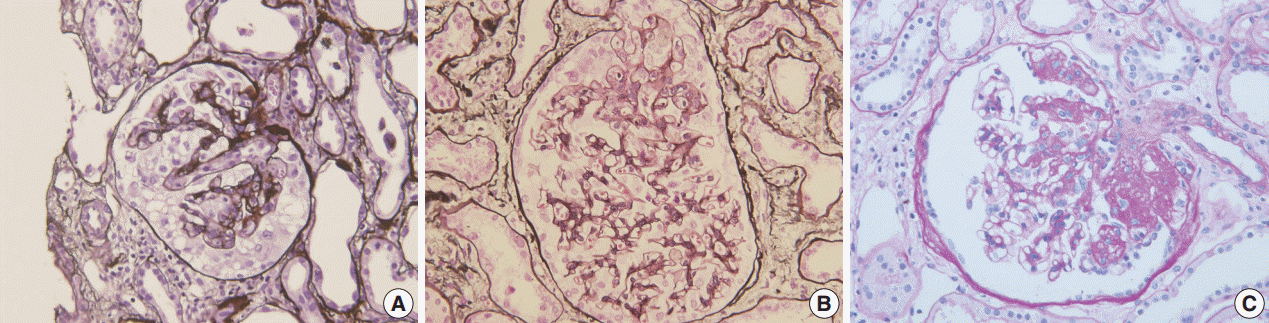
Morphologic variants of focal segmental glomerulosclerosis. (A) Capillary collapse and podocyte hyperplasia are characteristic features of the collapsing variant. (B) In the tip variant, segmental lesion involves the glomerular tuft next to the tubular pole. (C) The perihilar variant is diagnosed when sclerosis or hyalinosis are present in perihilar lesion in more than half of the sclerotic glomeruli.
PATHOGENESIS OF PODOCYTE INJURY
There are several observations indicating that podocyte injury is at the center of the development of FSGS. First, podocyte injury is the earliest morphologic feature of FSGS. In recurrent FSGS in the allograft kidney, podocyte injury is detected by electron microscopy prior to the development of overt sclerosis [14-16]. Second, there are animal models of podocyte-specific injury resulting in FSGS. NEP25 mice express human CD25 specifically on podocytes. Injection of immunotoxin which binds to human CD25 induced podocyte-specific injury and FSGS occurred a few weeks later [17]. Rats expressing diphtheria toxin receptors on podocytes developed FSGS after diphtheria toxin injection [18]. Third, histologic appearance of FSGS and clinical symptoms were in proportion with the number of injured podocytes [18]. Therefore, the pathogenesis of podocyte injury is a key to understand the characteristics of FSGS.
CIRCULATING PERMEABILITY FACTORS
As for the pathophysiology of podocyte injury, several mechanisms have been proposed with supporting evidences. Circulating permeability factors have been reckoned as the initiating factor of podocyte injury in primary FSGS and its recurrence after transplantation. The presence of serum factors that can cause podocyte injury was suggested from the therapeutic effect of immunoadsorption therapy [19] and observations that plasmapheresis could decrease the glomerular injury induced by patients’ serum [20]. Further, serum of recurrent FSGS patients significantly increased albumin permeability of glomeruli in an in vitro test [21]. Among the proposed circulating permeability factors, soluble urokinase receptor (suPAR) has been most thoroughly investigated. Wei et al. [22] presented data in a mouse model suggesting that the urokinase receptor of podocytes contributed to podocyte loss and proteinuria. The same group also suggested that suPAR could be the cause of FSGS. Serum levels of suPAR were increased in about two-thirds of primary FSGS patients and were also associated with recurrent FSGS after transplantation [23]. They also demonstrated that suPAR was increased in two different cohorts of biopsyproven FSGS patients. However, suPAR was inversely correlated with estimated glomerular filtration rate (eGFR) and treatment response [24]. Other authors found that plasma suPAR level was significantly increased in FSGS patients versus patients with minimal change disease, membranous nephropathy, or normal control. However, suPAR level was not useful in distinguishing primary and secondary FSGS [25]. Several contradicting reports on suPAR have also been published. Importantly, eGFR affects plasma levels of suPAR in patients with non-FSGS glomerular lesions, and a suPAR cut off value could not be determined even in FSGS patients due to the effect of eGFR [26]. Plasma levels of suPAR were also increased in lupus nephritis patients compared to lupus patients without renal involvement [27]. In IgA nephropathy patients, the plasma level of suPAR was related to the development of secondary segmental sclerosis [28,29]. Therefore, whether suPAR plays a role in the development of focal segmental lesions and its specificity to the primary FSGS are still open to further investigation.
Cardiotrophin-like cytokine-1 (CLC-1 or cardiotrophin-like cytokine factor 1 [CLCF-1]) is another candidate circulating permeability factor for primary FSGS. Savin et al. [30] have published on a serum factor purified from FSGS patients, which increased albumin permeability in isolated rat glomeruli. This factor had affinity for galactose and its molecular weight was less than 30 kDa. They identified this factor as CLC-1 by proteomic analysis and also found that the activity of CLC-1 was decreased by several factors such as heterodimer formation with cosecreted cytokine receptor-like factor 1 (CRLF1), Janus kinase 2 (JAK2) inhibitor, and signal transducer and activator of transcription 3 (STAT3) inhibitor [31,32]. A phase II clinical trial on therapeutic effect of galactose in patients with steroid-resistant FSGS was performed with inconclusive results due to small sample size [33,34]. This study design is interesting, considering that CLC-1 has high affinity for galactose and the CLC-1–galacotse complex can be easily removed in the liver [30].
Though known to be related to minimal change disease rather than FSGS, angiopoietin-like-4 (Angptl4) is also of interest [35]. Angptl4 has different functions according to its sialylation. While proteinuria was induced by hyposialylated Angptl4 located within the glomerulus, normosialylated Angptl4 was present in the peripheral circulation and mediated hypertriglyceridemia [35,36], indicating that these two symptoms of nephrotic syndrome could be linked through a common circulating factor.
GENETIC BACKGROUND
FSGS, as a podocytopathy, may be caused by mutation in several genes, which are important in maintaining podocyte morphology and function. Most of these genes can be categorized as those which are related with slit diaphragm structure, actin cytoskeleton of podocytes, or podocyte-glomerular basement membrane interaction through foot processes [37-39]. In addition, a specific channel mutation (see below) has also been identified as a cause of FSGS (Table 1). Alteration of these genes results in autosomal dominant or recessive congenital, infantile, or late onset nephrotic syndrome, some of which presents as FSGS histologically [40]. Mutation in the NPHS1 gene and resulting loss of its product nephrin, are responsible for congenital nephrotic syndrome of Finnish type. The locus of NPHS1 was identified at 19q13.1 in 1998 [41], which was the first identification of a podocytopathy-related gene. After this discovery, NPHS2 [42,43], PLCE1 (phospholipase Cε1, NPHS3) [43], WT1 (Wilms tumor 1, NPHS4) [44], LAMB2 (laminin β2, NPHS5) [45], PTPRO (protein tyrosine phosphatase receptor type O, NPHS6) [46], ARHGDIA (Rho GDP dissociation inhibitor α, NPHS8) [47], ADCK4 (aarF domain containing kinase 4, NPHS9) [48], and EMP2 (epithelial membrane protein 2, NPHS10) [49] were identified and related to autosomal recessive nephrotic syndrome. Many other genes related to nephrotic syndrome have been identified including ACTN4 (actinin α4, FSGS1) [50], TRPC6 (transient receptor potential cation channel 6, FSGS2) [51], CD2AP (CD2-associated protein, FSGS3) [52], APOL1 (apolipoprotein L1, FSGS4) [53], INF2 (inverted formin, FSGS5) [54], MYO1E (myosin 1E, FSGS6) [55], PAX2 (paired box gene 2, FSGS7) [56], ANLN (anillin, FSGS8) [57], and CRB2 (Crumbs homolog 2, FSGS9) [58]. There are interactions of these genes and their products. For example, WT1 transcriptionally regulates nephrin encoding of NPHS1, therefore, WT1 mutations influence NPHS1 function [59]. A study in a European cohort reported that two thirds of nephrotic syndrome within 1 year of life are related to alteration of NPHS1, NPHS2, WT1, or LAMB2 [60]. Another study in a non-Finnish ethnic group also reported that NPHS1 and NPHS2 mutations were the most common genetic alterations in congenital nephrotic syndrome [61]. In contrast to these Western studies, a genetic analysis of 30 Korean congenital and infantile nephrotic syndrome patients revealed that WT1 and NPHS1 mutations were the most frequent alterations, while NPHS2 mutations were the lowest frequency genetic alteration [62].

Genes related to FSGS or nephrotic syndrome
PARIETAL EPITHELIAL CELLS AND PODOCYTE INJURY
Podocytes are terminally differentiated cells having very limited ability of regeneration or proliferation. Therefore, the mechanism of repopulation of podocytes after podocyte injury has been of great interest. Recently, it has been suggested that parietal epithelial cells (PECs) lining Bowman’s capsule play an important role in this process by migrating from their original site to replace injured podocytes [63]. During glomerulogenesis, PECs and podocytes originate from common mesenchymal progenitors and finally have different phenotypes. Although little is known about the function of terminally differentiated PECs, they express tight junction molecules such as claudin-1, zonula occludens-1, and occludin and have barrier function against protein [64]. Some PECs express both CD133 and CD24, which are known to be stem cell markers, and these cells have regenerative ability [65]. More detailed study revealed that PECs show hierarchical differentiation according to their locations. PECs located at the urinary pole express CD133 and CD24 without the expression of podocyte markers (nestin, complement receptor-1, and podocalyxin). PECs of the vascular pole express podocyte markers without the expression of CD133 or CD24. In other areas, PECs express both CD133/CD24 and podocyte markers [66]. CD133 and CD24-expressing PECs have the ability to ameliorate kidney injury by potentiating tubular regeneration [65] and podocyte replacement, however, they can also contribute to glomerular injury such as glomerulosclerosis and crescent formation [67,68]. Animal models and human posttransplant biopsies demonstrated that invasion of activated PECs through the adhesion sites of the capillary tuft contributed to the development of FSGS [69]. The adhesion of the glomerular tuft to the Bowman’s capsule as a bridge of PEC migration appears to occur at early stages of FSGS development [70]. Therefore, detecting activated PECs on Bowman’s capsule or on the glomerular tuft could be an adjunctive diagnostic tool for early FSGS. In support of this concept, CD44 as a marker of activated PECs successfully distinguished early primary FSGS [70] and early post-transplant recurrence of FSGS [71] from minimal change disease. Interestingly, mutation of ARHGDIA, which is responsible for nephrotic syndrome, increased migration activity of cultured podocytes [47].
CONCLUSION
The etiology and pathogenesis of FSGS are very complex. Current research is focusing on the role of podocytes and interaction with PECs. Understanding the mechanism of podocyte injury, its progression and possible recovery is important not only for basic research but also for daily diagnostic pathology practice.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.