Alveolar Rhabdomyosarcoma of the Lip in an Adult with Clear Cell Features
Article information
Rhabdomyosarcoma, which occurs in the head and neck, genitourinary tract and deep soft tissue of the extremities and typically presents in childhood or adolescence, is a malignant mesenchymal tumor showing differentiation of skeletal muscle [1,2]. Less than 3% of rhabdomyosarcoma cases present in adults. Although rhabdomyosarcoma frequently occurs in the head and neck region, involvement of the lips is very rare; only 8 cases have been reported in the English literature [3-10]. Conventionally, rhabdomyosarcoma is categorized into embryonal, alveolar and pleomorphic subtypes [11]. We report a case of alveolar rhabdomyosarcoma with clear cell features presenting as a perioral subcutaneous nodule on the upper lip of a 58-year-old woman. A review of the literature and possible differential diagnoses are described.
CASE REPORT
A 58-year-old woman presented with a subcutaneous hard mass on her upper lip (Fig. 1A). A punch biopsy was performed at a local clinic and revealed small round cells infiltrating the dermis. The possibility of a small round cell tumor such as Merkel cell carcinoma or lymphoma was suggested. A wide excision was performed, and a skin-colored 0.7-cm nodule was noted. The cut surface of the mass was white gray and firm. Microscopic findings showed an ill-defined subcutaneous tumor that involved the entire dermis and subcutaneous adipose tissue sparing the epidermis (Fig. 1B, C). Small tumor cells infiltrated dissecting dermal collagen bundles and showed densely packed groups of cells. The cytoplasm was scanty with indistinct borders, and marked clear cell change was noted in most of the tumor cells (Fig. 1D). Some tumor cells had amphophilic cytoplasm. The nuclei were round to oval with focal indentation and finely granular chromatin. Frequent mitotic figures were noted up to 7 per 10 high power fields.
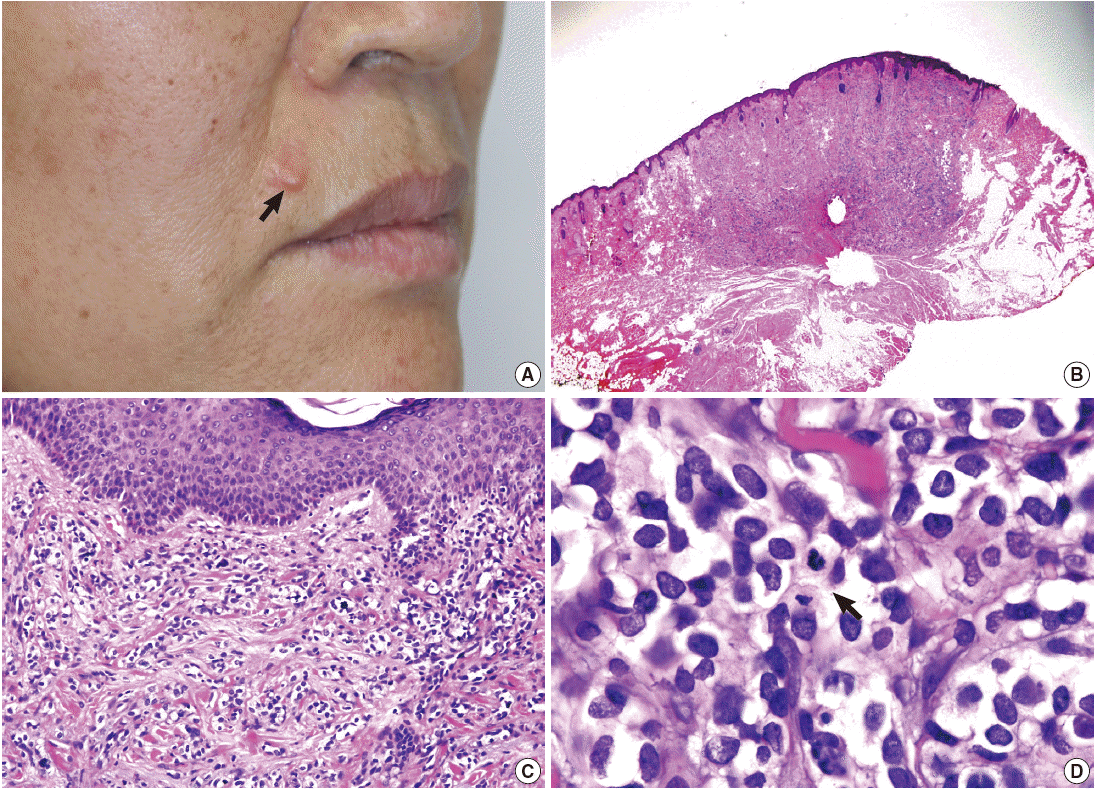
(A) A skin-colored nodule is noted in the upper lip (arrow). (B) An ill-defined mass involves the dermis and subcutaneous fat tissue. (C) The tumor cells extend to the papillary dermis but do not invade the epidermis. (D) The tumor is composed of densely packed groups of small round cells intersecting collagen bundles with marked clear cell change and frequent mitotic figures (arrow).
Periodic acid–Schiff stain demonstrated a few intracytoplasmic glycogen particles (Fig. 2A). Immunohistochemical examination of the tumor cells showed strong positivity for desmin, myogenin, and vimentin (Fig. 2B, C). The tumor cells were negative for cytokeratin, smooth muscle actin, human melanoma black 45, S-100 protein, and neuron-specific enolase. Under electron microscopy, the tumor cells showed thin and thick filaments with focal electron density in the cytoplasm, and many vacuoles which corresponds to clear cytoplasm visualized under light microscopy (Fig. 2D). These findings confirmed the diagnosis of rhabdomyosarcoma, alveolar type.

(A) Periodic acid–Schiff stain shows a few glycogen particles in the cytoplasm (arrow). (B) The cytoplasm is intensively immunoreactive for desmin, and a tumor cell shows striation-like features in the cytoplasm (asterisk). (C) The nuclei are positive for myogenin immunohistochemical stain. (D) Ultrastructurally, the tumor cell shows intracytoplasmic filaments (arrow) (×60,000).
After the diagnosis, head and neck computed tomography and positron emission tomography were performed and revealed no evidence of a primary tumor elsewhere or evidence of a mass in deeper soft tissue. The patient underwent radiation therapy and was free of recurrence or metastasis for four months after surgery.
DISCUSSION
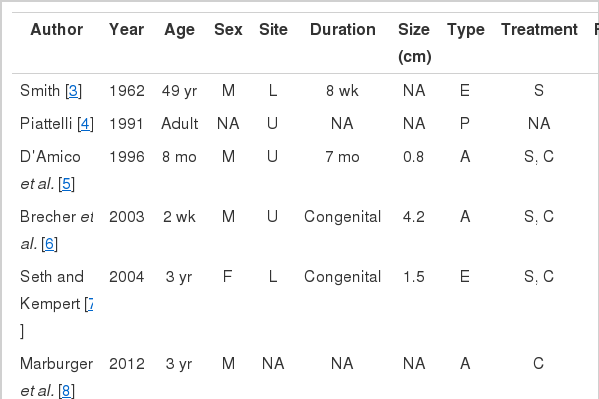
Rhabdomyosarcoma arising in the lip is very rare. Only eight cases have been reported in the English literature (Table 1) [3-10], and the patients ranged in age from 1 to 58 years. Three of nine patients including the current case were adults, and four of nine patients were male. Six patients had a mass in the upper lip. Three of nine patients presented with a congenital mass. The duration of symptoms ranged up to seven months, and the tumor size ranged from 0.7 cm to 10 cm. Five patients were diagnosed as alveolar subtype, three patients as embryonal subtype, and one patient as pleomorphic subtype. The follow-up period ranged from 3 to 71 months. None of the nine patients experienced local recurrence, but two had metastatic lesions. One patient had metastatic lesions at the cervical lymph node and salivary gland. The other patient had metastatic lesions at the lung and bone, who died of the disease within three months.
Clinicopathologic features of rhabdomyosarcoma of the lip
Rhabdomyosarcoma of the lip as a primary lesion is rare. The previously reported cases showed various clinical features such as ulcerated hemorrhagic soft mass, smooth mobile nodule, firm lobulated erythematous mass, lobulated small raised lesion, painful swelling and tender firm indurated mass. The clinical differential diagnoses include hemangioma, odontogenic tumor, dermoid, solitary fibromatosis, gingival granular cell tumor, neurofibroma and rhabdomyosarcoma. Microscopically, when the tumor shows skeletal muscle differentiation such as a rhabdoid feature, strap cells or cytoplasmic striation, it is easy to reach a diagnosis of rhabdomyosarcoma. However, in the case of a small biopsy or small tumor, the diagnosis may be more obscure. Several tumors including lymphoma, Ewing sarcoma and rhabdomyosarcoma can show the histologic features of a small round cell tumor. Suspicion of this diagnosis and an extensive search for the signs of differentiation on light microscopy are needed in such cases. An immunohistochemical study including desmin and myogenin is helpful if tumor cells do not show characteristic differentiation on light microscopy. Electron microscopy and genetic study are useful when the immunohistochemical staining is not diagnostic. Ultrastructural rhabdomyosarcoma tumor cells show cytoplasmic thick and thin filaments resembling rudimentary sarcomeric structure, cross striation, and glycogen particles.
Rhabdomyosarcoma with clear cell features has been rarely described in the English literature [12,13]. The clear or vacuolated cytoplasm visible on light microscopy is due to lipid droplets or glycogen particles on electron microscopy. Lipid-rich rhabdomyosarcoma resembles liposarcoma but the tumor cells are immunoreactive to muscle-specific markers such as desmin and myogenin. Variable amounts of glycogen can be observed in rhabdomyosarcoma and can be demonstrated with periodic acid–Schiff stain. The clear cell change itself obscures the definitive diagnosis of rhabdomyosarcoma especially when the clear cell features are dominant within the tumor. Tumors presenting as a dermal or subcutaneous nodule, which can show clear cytoplasm, are diverse and include balloon cell nevus, balloon cell melanoma, clear-cell sarcoma, Paget’s disease, clear-cell basal cell carcinoma, clear-cell syringoid tumors, clear-cell hidradenoma/hidradenocarcinoma, trichilemmoma/trichilemmocarcinoma, sebaceous neoplasm, lipoma/liposarcoma, cutaneous metastasis from renal cell carcinoma and clear-cell dermatofibroma [14].
Recently, a great advance in the ability to distinguish each of the subtypes of rhabdomyosarcoma has been demonstrated [11]. In alveolar rhabdomyosarcoma, reciprocal translocation of t(2;13) (q35;q14) and t(1;13)(p36;q14) and their associated fusions were identified. In embryonal rhabdomyosarcoma, chromosomal losses (chromosome 9 and 10) and gains (chromosome 8, 2, 7, 11, 12, 13, and 20) occur, as do allelic losses and mutations. Chimeric proteins from the fusion of PAX3 or PAX7 with FOXO1 are expressed in most alveolar rhabdomyosarcoma cases, resulting in the worse prognosis with this subtype.
Due to the rarity of this tumor, the diagnosis of adult rhabdomyosarcoma with clear cell features in the lip may be challenging in daily practice of surgical pathology. If the diagnosis of rhabdomyosarcoma is made in a lip biopsy, clinical investigation should be performed to eliminate the possibility of metastasis from other primary sites. Complete surgical excision and close follow-up are needed because of the possibility of metastasis.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.