The Utilization of Cytologic Fine-Needle Aspirates of Lung Cancer for Molecular Diagnostic Testing
Article information
Abstract
In this era of precision medicine, our understanding and knowledge of the molecular landscape associated with lung cancer pathogenesis continues to evolve. This information is being increasingly exploited to treat advanced stage lung cancer patients with tailored, targeted therapy. During the management of these patients, minimally invasive procedures to obtain samples for tissue diagnoses are desirable. Cytologic fine-needle aspirates are often utilized for this purpose and are important not only for rendering diagnoses to subtype patients’ lung cancers, but also for ascertaining molecular diagnostic information for treatment purposes. Thus, cytologic fine-needle aspirates must be utilized and triaged judiciously to achieve both objectives. In this review, strategies in utilizing fine-needle aspirates will be discussed in the context of our current understanding of the clinically actionable molecular aberrations underlying non-small cell lung cancer and the molecular assays applied to these samples in order to obtain treatment-relevant molecular diagnostic information.
During this current era of precision medicine, our current knowledge regarding the molecular drivers underlying cancer continues to evolve. Therefore, anatomic pathology specimens are becoming increasingly utilized for molecular diagnostic assays in order to detect clinically actionable genetic abnormalities in addition to routine diagnoses. This allows for tailored, personalized therapy in a subset of patients afflicted with malignancies harboring actionable genetic mutations for which approved targeted therapeutic agents exist. Molecular testing is therefore most relevant in the context of high-stage, surgically unresectable cancers. In this setting, minimally invasive small biopsy procedures are preferable in that they are associated with low risk of complications for the patient. Cytologic fine-needle aspiration (FNA) specimens are especially suitable, in this regard, as FNA procedures represent rapid, efficient, and minimally invasive means to sample superficial and deep lesions. Technological advances in imaging modalities and instruments such as computed tomography and endobronchial ultrasound (EBUS) have facilitated accurate and precise sampling of the latter. This constellation of circumstances poses unique challenges and opportunities for pathologists as FNA samples must be judiciously utilized, not only for rendering accurate cytologic diagnoses, but also effectively preserved and triaged for anticipated, relevant downstream molecular diagnostic assays.
In the past decade, significant strides have been made in further optimizing the use of small biopsy specimens and FNAs of non-small cell lung carcinomas (NSCLCs), especially adenocarcinomas and mixed histological subtypes with a component of adenocarcinomatous differentiation, for molecular studies [1]. One commonly held misconception is that FNAs are generally insufficient for the performance of molecular assays. However, for instance, we and others have observed and demonstrated that cellularity on the order of 100–500 cells are sufficient for DNA sequencing-based assays [2,3]. For fluorescence in-situ hybridization (FISH) assays, 100 analyzable tumor cell nuclei are generally sufficient [3].
Successfully integrating molecular ancillary techniques into routine cytology practice is essential in facilitating the appropriate management of advanced stage cancer patients by their oncologists [4,5]. The purpose of this review is to describe the approach to processing lung cancer FNA specimens at our institution, within the contexts of our current understanding of clinically actionable genetic abnormalities in NSCLCs and diverse cytopreparatory platforms on which molecular diagnostic tests are performed.
FINE-NEEDLE ASPIRATION SPECIMEN PROCESSING
FNAs represent an advantageous, effective means to obtain diagnostic cellular material as they can often sample a wide area of the target lesion, acquire tumor cells with lower contamination by background stromal connective tissue elements, and allow for immediate assessment during rapid on-site evaluation (ROSE). During the on-site assessment, a member of the cytopathology team is present and prepares direct smears, using the contents expelled from the needle, at the location of the procedure. This is advantageous for three reasons: each needle pass can be examined to determine tumor cell adequacy; there is an opportunity to engage the clinical care provider in a conversation regarding the preliminary diagnosis and relevant molecular diagnostic tests; and the cytopathology team member can help ensure that the specimen is processed in a manner that optimizes judicious triage for ancillary tests, including molecular studies [4,5]. FNAs can be performed without ROSE and in this setting, the contents of the FNA needles are typically expelled and rinsed in a cell preservative solution for use in liquid-based cytology (LBC) preparations. Examples of LBC preparations include ThinPrep (Hologic, Bedford, MA, USA), SurePath (Becton Dickinson, Franklin Lakes, NJ, USA), thin-layer advanced cytology assay system (TACAS, MBL, Tokyo, Japan), and Liqui-PREP (LGM International Inc., Ft. Lauderdale, FL, USA). Nonetheless, ROSE is an effective means to maximize the chances of success in acquiring adequate tumor cells for diagnosis and anticipated molecular studies. The ultimate goal is to prevent unnecessary repeat procedures to obtain additional tissue just for molecular studies which can lead to delays in treatment.
For patients afflicted with lung cancer at our institution, we perform ROSE for cytopathologist-performed FNAs and for EBUS-guided FNAs, performed by our clinical colleagues. The contents of each needle pass are expelled onto a slide which is utilized to prepare direct smears. The needle is then rinsed into buffered media; we utilize RPMI media for this purpose. Commonly, a pair of direct smears is prepared per needle pass; one is air-dried and the other is immediately alcohol-fixed. The air-dried smear is stained on-site with the Diff-Quik (Romanowsky) stain and the stained slide can be examined under the microscope immediately thereafter. The alcohol-fixed smear is stained later in the cytopathology laboratory with the Papanicolaou stain. Alternatively, the needle contents can be distributed over multiple smears allowing for flexibility in the utilization of direct smears for cytomorphologic evaluation and ancillary studies (Fig. 1) [4,5]. Based on the findings in the Diff-Quik stained smears, the determination can be made to perform additional needle passes to obtain more tumor cells for diagnosis and/or anticipated ancillary studies while the patient is still accessible. For instance, additional direct smears can be prepared and/or dedicated needle passes for the rinse solution (i.e., the needle contents are rinsed in the RPMI solution without the preparation of smears for those needle passes) obtained for the cell block preparation. This overall approach is flexible, forgiving, and engages the cytopathology team in maximizing the chance of success in obtaining adequate cellular material for diagnosis and ancillary studies. This approach can help minimize the chances of encountering the scenario in which diagnostic cellular material is present on only one smear; microdissection of the cells for molecular studies can result in sacrificing this only diagnostic slide in this context, which can have medico-legal consequences [6]. If this scenario is encountered, however, this risk can be mitigated by digital archiving prior to microdissection either via digital slide scanning and/or obtaining photomicrographs [7]. After immediate assessment of the cytomorphologic findings on the Diff-Quik stained smears, a preliminary diagnosis can be rendered by the cytopathologist and communicated to the clinical care providers.
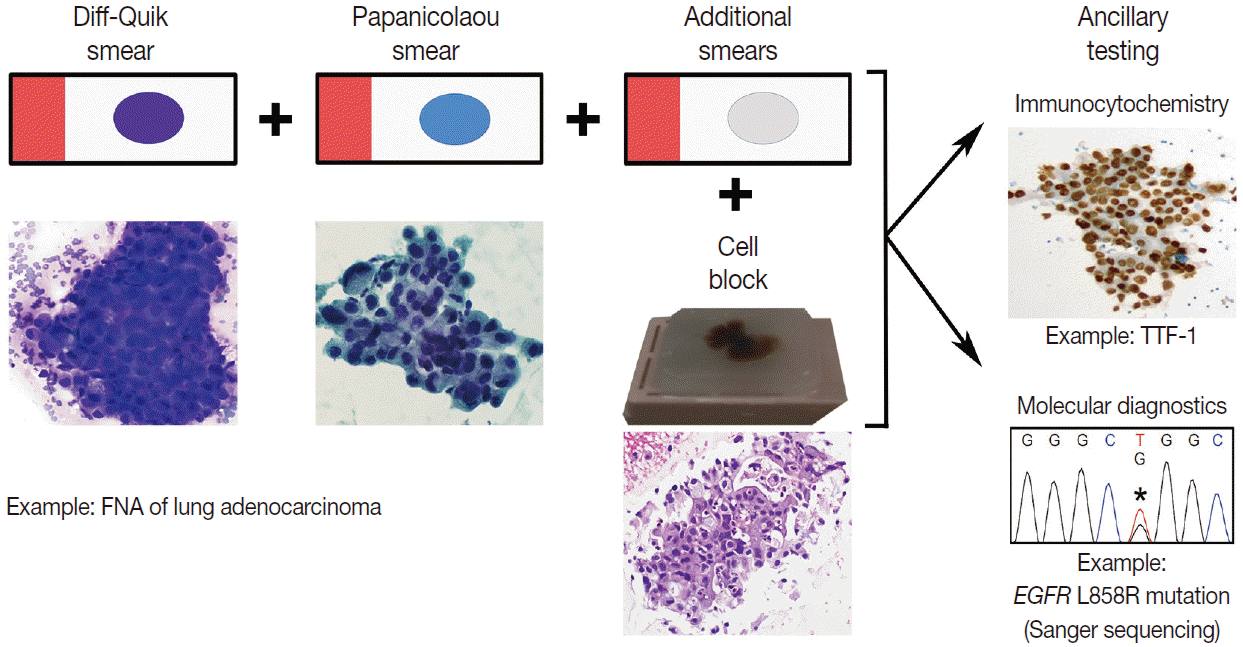
Example of fine-needle aspiration (FNA) processing workflow. The contents of a needle pass, obtained during an FNA procedure, are expelled onto a slide to prepare smears. Typically, a pair of smears (one Diff-Quik stained and one Papanicolaou stained) is prepared per needle pass. However, additional smears can be prepared from a single needle pass by distributing the cellular material across more than two slides. The additional smears can be directly triaged for ancillary studies. For instance, an unstained smear can be sent to the Immunohistochemistry Laboratory for immunostaining (e.g., thyroid transcription factor-1 [TTF-1] immunocytochemistry). Also, a stained smear can be directly triaged to the Molecular Diagnostics Laboratory for tumor cell macro- or microdissection, nucleic acid isolation, and subsequent molecular testing (e.g., epidermal growth factor receptor [EGFR] mutation analysis).
After the conclusion of the procedure, the needle rinse solution, containing a suspension of aspirated cells, is centrifuged in the cytopathology laboratory to pellet the cells. Once the supernatant is removed, the cell pellet is congealed in a matrix; an agar-like substance such as HistoGel (Thermo Scientific, Waltham, MA, USA) can be used or the cells can be mixed with plasma and thrombin to create a clot. At our institution, we employ HistoGel for this purpose. The resulting cell button is then placed in a cassette. Commonly, this cassette is fixed in formalin and processed to ultimately create a formalin-fixed, paraffin-embedded (FFPE) cell block. This is analogous to a FFPE block of small biopsy tissue, which can be sectioned for evaluation via the hematoxylin and eosin stain and ancillary tests such as immunocytochemistry or molecular diagnostic assays. It should be noted that heavy metal fixatives such as Zenker’s fixative and acidzinc-formalin, acidic fixatives such as Bouin’s solution, or decalcification solutions should generally be avoided as these render specimens unusable for molecular testing [3].
Cell blocks are traditionally utilized for ancillary immunocytochemistry and molecular diagnostic assays. The main advantage of using cell blocks is that the majority of ancillary tests are validated for FFPE sections; FFPE cell blocks are treated similarly to traditional surgical pathology FFPE blocks. Furthermore, multiple serial sections from cell blocks can be utilized to perform a battery of ancillary studies. There are two main disadvantages associated with cell blocks, however. First, adequate cellularity in cell blocks is not guaranteed at the time of the procedure. Performing dedicated needle passes, for the needle rinse solution during the FNA procedure, may improve the chances of obtaining a sufficiently cellular cell block but still does not guarantee success [4]. In addition, for hypocellular cell blocks, there is a risk of depleting the cells of interest upon deeper sectioning for molecular tests. Second, the cell block is derived from a needle rinse solution which is a pooled specimen that contains contents of all the needle passes. Thus, if one or more needle passes contain tumor cells but other needle passes contain abundant benign background cellular elements, the tumor cells will be diluted by these benign cells in the cell block [4]. In addition to the above disadvantages, DNA extracted from FFPE cell blocks may produce sequencing artifacts as formalin fixation leads to the crosslinking of nucleic acids and proteins and the possibility of sequence alterations [8].
Recently, given the shortcomings of cell blocks, there has been an increasing appreciation of alternative cytopreparatory platforms for molecular testing such as direct smears and LBC samples. In the subsequent sections below, the utilization of these cytopreparatory platforms will be discussed in the context of the clinically relevant mutations and translocations analyzed during molecular diagnostic testing of NSCLCs.
EPIDERMAL GROWTH FACTOR RECEPTOR
The epidermal growth factor receptor (EGFR) gene encodes a transmembrane growth factor receptor that exhibits tyrosine kinase activity. Upon activation, intracellular signaling is mediated by cytoplasmic effectors in the RAS-RAF-MEK-ERK, PI3K-AKT, and STAT pathways [8]. Approximately 10%–15% of NSCLC harbor clinically relevant, sensitizing mutations in EGFR. EGFR mutations are predominantly observed in lung adenocarcinomas; the L858R substitution and small in-frame deletions in exon 19 are the most commonly observed mutations and account for up to 90% of all EGFR mutations in this setting [3,9]. These mutations are more commonly associated with East Asian ethnicity, female gender, and non-smoking history. However, these are not absolute rules and clinical characteristics should not be used to exclude lung cancer patients from mutation testing [10]. Lung adenocarcinomas harboring these sensitizing mutations in EGFR have been shown to respond to EGFR tyrosine kinase inhibitors (TKIs). The first-line TKIs include erlotinib and gefitinib. In clinical trials, TKI therapy has been demonstrated to result in improved progression-free survival, compared to standard chemotherapy, for patients with lung adenocarcinoma harboring EGFR mutations [11-16].
EGFR mutation analysis is commonly performed via polymerase chain reaction (PCR) and sequencing-based approaches; advances in the development of testing modalities have afforded a multitude of methodologies [1,17,18]. Sanger sequencing is considered the gold standard as this involves direct DNA sequence acquisition and can provide information regarding the presence of all potential mutations including common, known mutations and novel mutations. Nonetheless, this test requires a relatively higher enrichment of tumor cell DNA content in the sample. The typical analytic sensitivity for Sanger sequencing is 15%–20% mutant allele, which equates to 30%–40% tumor cells assuming that the genetic mutation is a heterozygous event without amplification [17]. This can be problematic in both small biopsy and cytology specimens, especially cell blocks, in which the tumor cell population can be diluted by background benign cellular elements such as inflammatory cells, bronchial epithelial cells, and/or stromal mesenchymal cells. Especially in this setting, a negative mutation result can be either due to the true absence of the mutation in the tumor cells or insufficient percent tumor cellularity that falls below the analytic sensitivity threshold thereby resulting in the failure to detect the mutation even despite the presence of the mutation [4,9,17,19]. Therefore, often times, there is more reliance on tumor cell enrichment by either macrodissection or microdissection to obtain a reliable result [7,9,17,20]. Sanger sequencing is also relatively more labor intensive and time consuming than targeted methods and can lead to longer turnaround times [18]. In contrast to the general Sanger sequencing approach, targeted mutation detection methods such as PCR-restriction fragment length polymorphism, real-time PCR, pyrosequencing, high resolution melting analysis (HRMA), and PCR fragment analysis can be utilized [17]. The advantages of these approaches include their improved analytic sensitivity and less time-consuming nature leading to reduced turnaround times. At our institution, we utilize a multiplex PCR fragment analysis assay for EGFR mutation testing; this allows for the simultaneous assessment of the two most commonly observed EGFR mutations (Fig. 2). The analytic sensitivity of this method is better than that of Sanger sequencing; a minimum of only 10% tumor cells is required. In the past decade, myriad studies have been reported demonstrating that a variety of cytologic samples and cytopreparatory platforms can be effectively utilized for EGFR mutational analysis. These have been reviewed elsewhere [18,21] but salient examples will be discussed below.
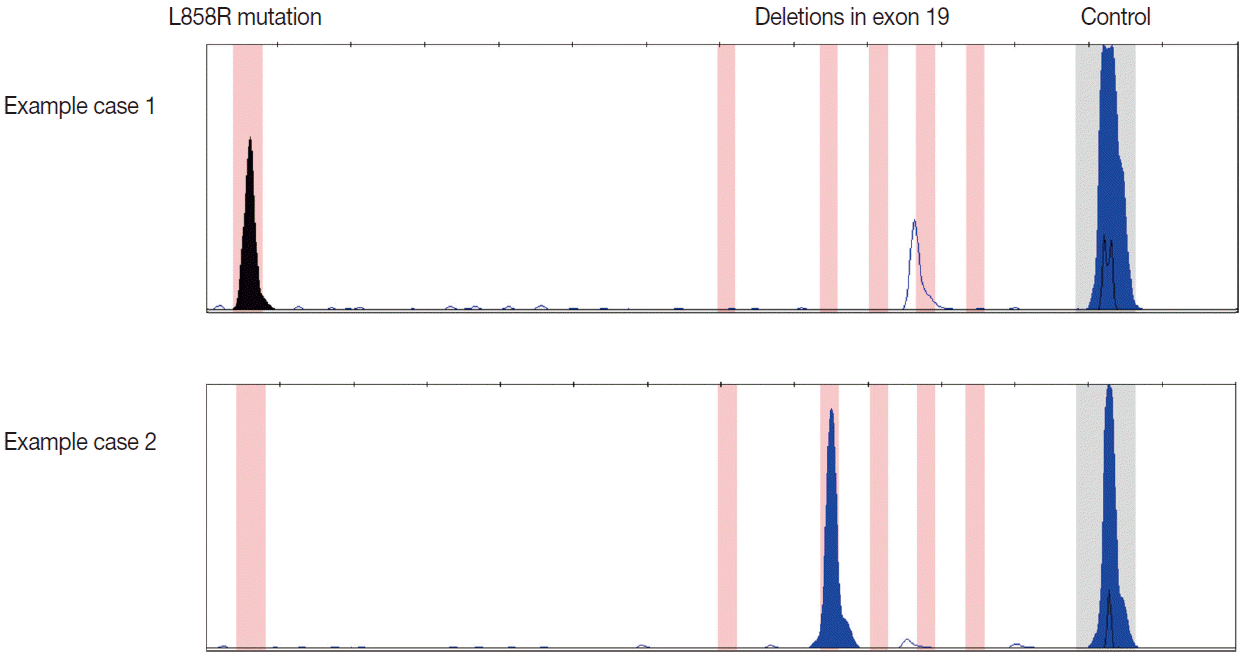
Examples of epidermal growth factor receptor (EGFR) mutations detected by the polymerase chain reaction based fragment analysis assay utilized at our institution. Our assay is a multiplex assay designed to detect the two most common mutations in EGFR: the L858R substitution (case 1) and small deletions within exon 19 (case 2).
As mentioned previously, FFPE cell blocks represent the traditional cytopreparatory platform on which ancillary molecular diagnostic tests are performed. Much of the reasoning behind this lies in that FFPE cell blocks best approximate FFPE blocks containing tissue specimens and the majority of the molecular assays are validated using FFPE material. Nonetheless, in light of the inherent disadvantages of cell blocks mentioned previously, investigation into other cytopreparatory platforms for molecular diagnostic assays have been performed by our group and many others. Cytopathology specimen preparation is diverse and versatile; the consequence of this is that each type of cytologic analyte platform needs to be carefully validated for any given molecular test. Cytologic direct smears may ultimately prove to be the cytologic platform best suited for PCR-based analysis due to the high quality of nucleic acids and immediate nature of specimen assessment for tumor cell adequacy [4,17]. Cells on direct smears are not exposed to formalin but rather undergo an alcohol-based fixation process prior to staining; thus, the negative effects of formalin fixation on nucleic acid quality is non-contributory for direct smears. Furthermore, the tumor cells of interest can be directly visualized on the direct smear and isolated for nucleic acid extraction and molecular analysis; in essence, “what you see is what you get.” We have previously demonstrated that uncoverslipped and previously coverslipped/decoverslipped Diff-Quik stained smears are robust sources of cellular material for PCR-based mutational analysis such as EGFR mutation testing of lung adenocarcinoma and BRAF mutation testing of metastatic melanoma FNA specimens [9,19,22]. We and others have observed that destaining the smears is not necessary for successful DNA extraction and PCR [9,21]. Our preference in utilizing Diff-Quik stained smears over Papanicolaou stained smears is rooted in the following observations: (1) cellular material on a Diff-Quik stained smear can be easily visualized without a coverslip and immediately triaged for molecular diagnostic testing [4]; and (2) the report by Killian et al. [23] that nucleic acids extracted from Diff-Quik stained smears show better preservation and integrity than DNA extracted from Papanicolaou stained smears. With respect to the latter point, others have pointed out that Papanicolaou stained smears are just as feasible for PCR-based molecular tests [7,21,24]. Multiple groups have successfully demonstrated the utilization of cytologic direct smears for EGFR mutation testing of lung adenocarcinomas [2,9,25-36] with low failure rates and high degree of concordance with EGFR molecular testing results from corresponding histologic specimens, when available. In our experience, 100 to 200 tumor cells are sufficient for successful DNA isolation and EGFR mutation analysis and this is congruent with the observations from others [2,21].
LBC samples have also been investigated for EGFR mutation testing and have been shown to be an effective analyte for this purpose. This is valuable when a member of the cytopathology team is not available for ROSE [37]. Several LBC technologies have been developed including ThinPrep, SurePath, TACAS, and Liqui-PREP. Of these, the emerging literature, to date, on the utilization of LBC samples for molecular testing of NSCLC have predominantly focused on Cytolyt (Cytyc Corp., Marlborough, MA, USA) cell suspensions and ThinPrep slides. Collecting FNA samples in the methanol-based Cytolyt solution results in reduction of background blood as the Cytolyt solution exhibits hemolytic properties [7]. The cell suspension can be divided and an aliquot is used to prepare a ThinPrep slide that is stained via the Papanicolaou method, similar to alcohol-fixed Papanicolaou stained direct smears. Tumor cells can be isolated from the ThinPrep slide and used for DNA isolation and EGFR mutation analysis [38,39]. Furthermore, the cell suspension aliquot that is not used to prepare a ThinPrep slide can be centrifuged and the cell pellet itself can be used as an analyte for mutation testing [38-40]. Similar to the needle rinse cell suspension utilized to prepare cell blocks, the Cytolyt cell suspension is a pooled specimen if multiple needle passes are expelled and rinsed into Cytolyt. Thus, if one or more needle passes are high in tumor cell content whereas other needle passes contain a high proportion of benign cellular elements, the tumor cells in the final pooled cell suspension will be diluted [4]. This is not necessarily an insurmountable challenge for two reasons. First, targeted methods for detecting EGFR mutations with improved analytic sensitivity, relative to Sanger sequencing, can be utilized [38]. Second, tumor cell enrichment from the ThinPrep slide can be accomplished by laser capture microdissection (LCM). To illustrate these points, Malapelle et al. [39] directly compared the performance of EGFR mutation analysis by Sanger sequencing using paired samples for each case analyzed: (1) pelleted cells from the Cytolyt suspension and (2) tumor cells obtained from the ThinPrep slide via LCM. They observed that EGFR mutations were more reliably identified in the latter rather than the former. This difference was minimized when utilizing more sensitive EGFR mutation analytic approaches which included HRMA and PCR fragment analysis assays [38]. Based on these studies, the authors speculated that coupling the use of highly sensitive EGFR mutation detection assays and Cytolyt derived cell pellets may be sufficient obviating the need for microscopy. Nonetheless, it must be emphasized that any negative molecular testing result should carefully be reconciled with the analyte input utilized for the mutation assay. This is essential to determine whether the negative result represents a true negative or potentially a false-negative, necessitating possible retesting. In this regard, the utilization of microscopy still remains an essential pre-analytic quality assurance activity to best ensure that the input analyte is of sufficient tumor cellularity to maximize confidence in the results of the mutation assay. Of note, a follow-up study by Bellevicine et al. [6] compared the utility of direct smears versus ThinPrep slides. They observed that the direct smears exhibited significantly higher cellularity than ThinPrep slides, on average. Accordingly, the average yield of DNA extracted from direct smears was significantly higher than that from ThinPrep slides.
As the L858R substitution and deletions in exon 19 represent approximately 90% of all EGFR mutations in lung adenocarcinoma [41], rabbit monoclonal antibodies have been recently developed and investigated. One antibody is specific for the exon 21 L858R mutation and the other specific for the 15-base pair/5-amino acid deletion (E746_A750del) in exon 19 (clone 6B6). The immunohistochemical approach to detecting mutant EGFR proteins using these antibodies has been examined in several studies on lung cancer tissues as well as cytologic and small biopsy samples [42-48]. The sensitivity of these assays range from 47%–92% but their high positive predictive value and specificity supports the feasibility of utilizing this approach as a first-line screening approach [3]. Two cautionary points are worth mentioning with regards to this approach. First, immunohistochemistry should be performed after careful validation and formulation of immunostain scoring criteria. The significance of how best to interpret equivocal staining results should be clarified as overinterpreting weak immunoreactivity as a positive mutation result can lead to increased false positives and decreased specificity. Second, the clone 6B6 antibody best detects the 15-base pair (E746_A750del) EGFR deletion mutant protein but demonstrates variable immunoreactivity for the EGFR mutant proteins resulting from non-15-base pair deletions in exon 19 [43].
ANAPLASTIC LYMPHOMA KINASE
The anaplastic lymphoma kinase (ALK) protein is a receptor tyrosine kinase and rearrangements involving the ALK gene locus is observed in approximately 5% of lung adenocarcinomas [49,50]. These mutations are more commonly observed in younger age patients who are never-smokers. However, there can be exceptions to this and clinical characteristics should not be used to exclude lung cancer patients for ALK rearrangement testing [10]. Most ALK rearrangements in lung adenocarcinoma result from interstitial deletions and small inversions within the short arm of chromosome 2. This results in the fusion of portions of the echinoderm microtubule-associated protein-like 4 (EML4) and ALK genes [50,51]. The fusion gene product displays oncogenic activity that drives these ALK rearranged lung adenocarcinomas [50]. Other less common ALK rearrangements involve fusions between ALK and other genes, such as KIF5B and TFG [10]. ALK rearranged lung adenocarcinoma has been recognized as a legitimate target for small molecular inhibitor therapy; crizotinib was shown to be efficacious in treating patients with these cancers. In a phase 1 study evaluating 143 patients, an overall response rate of 61% and estimated overall survival rates of 74.8% at 12 months were observed [52]. Therefore, the evaluation of lung adenocarcinoma FNA samples for ALK rearrangements, in addition to EGFR mutations, has become increasingly incorporated into patient management algorithms.
FISH is currently the preferred approach to assaying lung adenocarcinomas for ALK rearrangements according to expert recommendations based on review of the literature [10]. In the United States of America, there is only one test approved by the Food and Drug Administration (FDA) for this purpose. This assay utilizes a dual ALK breakapart probe strategy in which orange and green labeled probes hybridize to the highly conserved translocation breakpoint region in the ALK gene. ALK gene loci that have not undergone rearrangement typically display fused orange and green signals (yellow) or juxtaposed touching orange and green signals. When an ALK rearrangement occurs, the orange and green signals become separated. However, as the majority of the ALK rearrangements involve a small inversion within chromosome 2p, rather than a rearrangement involving another chromosome, the extent to which the two signals are split is finite. In order to score a nucleus as positive for the ALK rearrangement, the orange and green signals must be separated by a distance of >2 signal diameters (Fig. 3); a nucleus can also be scored positive if a single orange signal without a corresponding green signal is observed [53]. Typically, up to 100 tumor cell nuclei are scored in this assay and a lung adenocarcinoma is considered positive for the ALK rearrangement if at least 15% of the nuclei are scored as positive for the rearrangement [53,54].
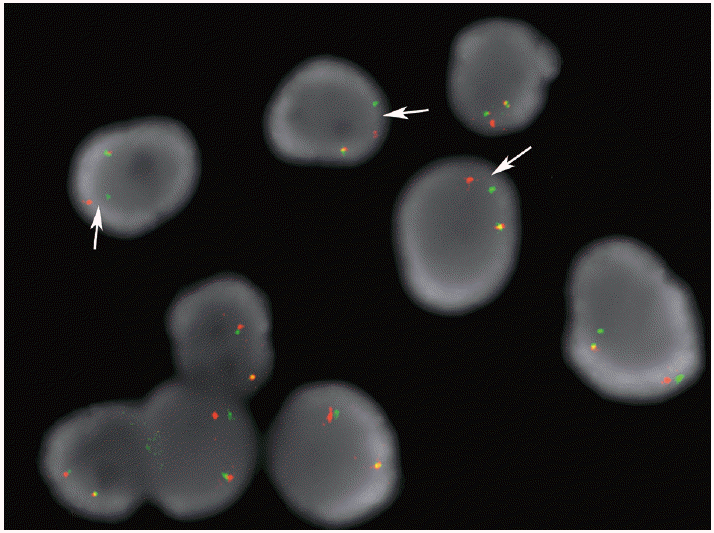
Example case of anaplastic lymphoma kinase (ALK) rearrangement fluorescence in-situ hybridization assay performed on a direct smear containing lung adenocarcinoma cells. The arrows point to the split orange and green signals that are separated by a distance of > 2 signal diameters. These nuclei would be scored as positive for the ALK rearrangement.
The FDA has approved this dual breakapart probe set for use on paraffin embedded tissue sections. As mentioned previously, paraffin embedded cell blocks are processed similarly to paraffin embedded tissue blocks; thus, cell blocks are traditionally used for the performance of ALK rearrangement FISH assays [55]. Nonetheless, there is an observed noticeable failure rate when using and relying on cell blocks for these assays due to insufficient tumor cell material in a significant proportion of cases [4,54]. Thus, we and others have investigated alternative cytopreparatory platforms for ALK FISH. In our study, we demonstrated the effective use of Diff-Quik stained direct smears for this purpose and observed that the performance of this assay using direct smears was better than the performance using cell blocks [53]. Proietti et al. [56] also examined the use of Papanicolaou stained direct smears and observed that the failure rate due to insufficient cellularity was significantly higher for cell blocks than for direct smears. Therefore, the utilization of both Diff-Quik and Papanicolaou stained smears are feasible analytes for ALK FISH. Recently, ThinPrep slides have also been shown to be a feasible platform for ALK FISH [54]. The advantage of utilizing cytologic preparation platforms such as direct smears and ThinPrep slides for this purpose is that entire tumor cell nuclei are being analyzed. FISH evaluation on paraffin sections derived cell blocks and other FFPE blocks are prone to signal loss in some of the tumor cells due to section truncation artifacts [53,54].
In addition, alternative methods to FISH have been the subject of recent investigation. Immunohistochemistry utilizing antibodies directed against ALK is an attractive alternative as it is simpler, quicker, and less expensive [10]. The challenge associated with this approach is that the ALK protein is expressed at much lower levels in ALK rearranged lung tumors than in anaplastic large cell lymphoma, the prototypical ALK rearranged tumor [10]. Fortunately, monoclonal anti-ALK antibodies (clones D5F3, D9E4, and 5A4) have been shown to exhibit high sensitivity and specificity [10,57]. Immunohistochemistry using the D5F3 and 5A4 monoclonal antibodies on cytologic specimens have been recently described and shown to exhibit a high degree of concordance with ALK FISH testing [54,58]. This supports the feasibility of utilizing an immunohistochemical approach as a first-line screening methodology to select specimens for ALK FISH testing. Of note, the mouse monoclonal anti-ALK1 antibody (CD246) typically used for the diagnosis of anaplastic large cell lymphoma is less reliable for identifying ALK rearrangements in lung adenocarcinoma; this is most likely attributable to the limited sensitivity of this particular antibody in detecting the lower expression levels of the ALK fusion proteins in lung adenocarcinoma relative to anaplastic large cell lymphoma [10]. Finally, the application of reverse transcriptase PCR (RT-PCR) to cytologic direct smears for ALK rearrangement analysis has been recently reported [31]. When evaluating a cohort of paired cytologic-histologic specimens by RT-PCR, Mitiushkina and colleagues observed a 100% concordance of results. Nonetheless, as a cautionary note, there is a high degree of variability in EML4-ALK fusion events with at least 13 variants of EML4-ALK being reported [10]. Furthermore, other fusion partners to ALK such as TFG and KIF5B can be observed. Therefore, an RT-PCR approach may not capture all clinically relevant ALK rearrangements [10].
EMERGING MOLECULAR GENETIC BIOMARKERS
While EGFR mutations and ALK rearrangements represent the two best characterized, clinically actionable molecular alterations in NSCLC, other molecular markers are becoming increasingly appreciated and investigated. ROS1 encodes a receptor tyrosine kinase and is rearranged in approximately 2% of lung adenocarcinomas [59]. Early data seem to indicate that these tumors are responsive to crizotinib. MET encodes another receptor tyrosine kinase, hepatocyte growth factor receptor, and is amplified in a subset of NSCLC; a significant proportion of these cases are seen in context of acquired resistance to EGFR TKIs [1]. As crizotinib also targets this receptor, studies are under way to determine whether this agent will be effective in MET amplified lung cancers. Next, mutations in BRAF are seen in approximately 3% of lung adenocarcinomas [60]. Therapeutic agents targeting BRAF, such as dabrafenib, are currently being investigated in clinical trials [61]. In addition, gene rearrangements involving the RET tyrosine kinase gene (e.g., KIF5B-RET) have been observed in approximately 2% of lung adenocarcinomas; RET specific TKIs such as sunitinib, sorafenib, and vandetanib may be useful in treating patients with these lung cancers [3]. Finally, amplification of FGFR1 and mutations in PIK3CA have been observed in some lung squamous cell carcinomas and agents targeting these gene products are also under investigation [1]. The above mentioned molecular genetic markers highlight the distinct molecular profiles that are becoming increasingly appreciated for different subtypes of NSCLC, especially adenocarcinomas and squamous cell carcinomas. Therefore, efforts to correctly subtype cases of NSCLC are important, including challenging cases of poorly-differentiated NSCLC [10]. At our institution, if the subtyping of NSCLC is deemed to be challenging based on cytomorphologic evaluation alone, immunocytochemistry is utilized; we have demonstrated that this can be accomplished using smears as well as cell blocks [4]. For example, Napsin-A and thyroid transcription factor-1 are often positive in lung adenocarcinomas and can be utilized for confirming this subtype of NSCLC. In contrast, p63 and p40 are useful markers for confirming a diagnosis of squamous cell carcinoma. Ultimately, utilizing FNA samples of NSCLC for immunocytochemistry must be judiciously leveraged to ensure that sufficient material still exists for molecular ancillary testing.
CONCLUDING THOUGHTS
Given the continuously evolving landscape in our understanding of the genetic events responsible for lung cancer pathogenesis, the integration between anatomic pathology, particularly cytopathology, and molecular diagnostics will become even more essential. Emerging molecular technological advances, such as next generation sequencing (NGS), that allow for high-throughput, high-sensitivity molecular analyses will likely play an important role in the management of patients with NSCLC. Cytologic specimens, based on recent reports, represent a robust source of cellular material for NGS [62-65].
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
I wish to thank my colleague in the Molecular Diagnostics Laboratory at University of Michigan Health System, Dr. Bryan Betz, for providing the images pertaining to molecular diagnostic assays displayed in this review.