Idiopathic Noncirrhotic Portal Hypertension: An Appraisal
Article information
Abstract
Idiopathic noncirrhotic portal hypertension is a poorly defined clinical condition of unknown etiology. Patients present with signs and symptoms of portal hypertension without evidence of cirrhosis. The disease course appears to be indolent and benign with an overall better outcome than cirrhosis, as long as the complications of portal hypertension are properly managed. This condition has been recognized in different parts of the world in diverse ethnic groups with variable risk factors, resulting in numerous terminologies and lack of standardized diagnostic criteria. Therefore, although the diagnosis of idiopathic noncirrhotic portal hypertension requires clinical exclusion of other conditions that can cause portal hypertension and histopathologic confirmation, this entity is under-recognized clinically as well as pathologically. Recent studies have demonstrated that variable histopathologic entities with different terms likely represent a histologic spectrum of a single entity of which obliterative portal venopathy might be an underlying pathogenesis. This perception calls for standardization of the nomenclature and formulation of widely accepted diagnostic criteria, which will facilitate easier recognition of this disorder and will highlight awareness of this entity.
Portal hypertension is a manifestation of increased resistance to portal venous flow due to prehepatic, intrahepatic, or posthepatic causes. The intrahepatic causes are further divided into presinusoidal, sinusoidal, and postsinusoidal causes. The two most common causes of portal hypertension are cirrhosis and schistosomiasis [1,2], both of which are intrahepatic. When no definitive cause for portal hypertension is identified in the absence of cirrhosis, the condition is referred as an idiopathic noncirrhotic portal hypertension (INCPH) [3].
Although INCPH has been recognized for more than one century, the knowledge about this disorder is relatively limited due to clinical and pathologic under-recognition and lack of standardized nomenclature and diagnostic criteria. Recent morphologic studies have greatly enhanced our understanding about INCPH by demonstrating its wide histologic spectrum and its common associations with other systemic conditions. Moreover, these studies have revealed that diverse morphologic entities of different names, such as hepatoportal sclerosis (HPS), partial nodular transformation (PNT), incomplete septal cirrhosis (ISC), and nodular regenerative hyperplasia (NRH), indeed represent temporal and spatial heterogeneity of a single condition whose underlying pathogenesis is obliterative portal venopathy (OPV) [4-7].
Recently, Schouten et al. [8] proposed INCPH as a unifying diagnostic terminology in order to encompass the variable histopathologic entities manifesting as portal hypertension in the absence of cirrhosis, with a clinical connotation in the terminology. Standardization of the nomenclature will not only promote optimal patient care, but will also facilitate sharing of knowledge and collaborative research, which will lead to development of guidelines for treatment and diagnostic criteria. This paper summarizes the historical background of INCPH including the evolution of the terminology and provides an updated review of its clinical and pathological aspects.
DEFINITION AND DIAGNOSIS
The Asian Pacific Association for the Study of the Liver (APASL) defines INCPH (referred as noncirrhotic portal fibrosis [NCPF]/idiopathic portal hypertension [IPH]) as “a disease of uncertain etiology characterized by a periportal fibrosis and involvement of small and medium branches of the portal vein, resulting in the development of portal hypertension [9].” In Western countries, the condition is poorly characterized and does not have a widely accepted definition, possibly due to its rarity. Generally, it is regarded as a clinical entity of intrahepatic portal hypertension with no evidence of cirrhosis, other liver diseases that might be accountable for portal hypertension, and splanchnic venous thrombosis [3,6,10,11].
The diagnosis of INCPH is rendered in patients with portal hypertension after excluding portal vein thrombosis, Budd-Chiari syndrome, exposure to medications or toxins, splenic vein thrombosis, and other liver disease that can manifest as portal hypertension, followed by a confirmatory liver biopsy [3]. Since INCPH is a diagnosis of exclusion, this poses challenges to clinicians and pathologists, and many patients carry a presumed diagnosis of cirrhosis [3,12].
HISTORY AND NOMENCLATURE
Between 1884 and 1910, Banti [13] described patients with splenomegaly and anemia without hematologic disorders. He speculated that the natural history of this disorder consisted of three phases—initial phase of splenomegaly and anemia, followed by a transitional phase, and finally progressing to a terminal phase of gastrointestinal hemorrhage, liver failure, and death [14]. In retrospect, his cohort was heterogeneous and included patients with cirrhosis, tropical splenomegaly due to malaria, and INCPH [8]. This “Banti’s syndrome” was thought to represent a primary splenic disorder with secondary changes in the liver, with endophlebitis as a common pathogenesis [15].
The paradigm shift occurred in 1934, when McMichael [16] attributed the pathologic changes of the portal veins to portal hypertension, in patients with “hepatolienal fibrosis,” i.e., splenomegaly without liver cirrhosis. In 1936, the Spleen Clinic at Columbia Presbyterian Hospital in New York reported 15 patients with splenomegaly with no evidence of cirrhosis or obstruction of the portal venous system [17]. Subsequently, in 1945, Whipple [18] reported that 26 of 93 patients with splenomegaly were without cirrhosis, schistosomiasis, or extrahepatic portal vein obstruction. The term “hepatoportal sclerosis (HPS)” was coined in 1965 by Mikkelsen et al. [19] for this condition. In their paper, the authors documented histologic evidence of phlebosclerosis of the intrahepatic and extrahepatic branches of the portal vein in 36 patients with noncirrhotic portal hypertension [19]. Phlebosclerosis was recognized as partial or complete obliteration of the portal vein lumen. Comparable histologic observation was reported in a study from Calcutta, India, wherein the authors used the term “idiopathic portal hypertension (IPH)” in the title and “noncirrhotic portal fibrosis (NCPF)” in the text in order to designate portal hypertension without cirrhosis or extrahepatic portal obstruction [20]. In addition, this study demonstrated a better prognosis of NCPF/IPH compared to that of cirrhosis. The term NCPF was subsequently endorsed by the Indian Council of Medical Research.
The pathogenic terminology “obliterative portal venopathy (OPV)” was introduced by Nayak and Ramalingaswami [21] in their pathologic study of noncirrhotic portal hypertension. OPV was characterized by segmental, conspicuous subendothelial thickening of large- and medium-sized intrahepatic portal vein branches. In addition, scarring and obliteration of small portal vein branches along with an increased number of small vascular channels within the portal tracts and incomplete thin fibrous septa were noted [21]. In Japan, the term IPH was used in a national survey performed by the Ministry of Health and Welfare [22]. Other names given to this disorder include noncirrhotic intrahepatic portal hypertension, benign intrahepatic portal hypertension, and idiopathic presinusoidal portal hypertension [6,14,23,24]. To date, the names OPV and HPS have been commonly used in the Western literature, and IPH and NCPF have been widely used in the Eastern regions. The unifying term INCPH was proposed in a review paper by Shouten et al [8], since this nomenclature addresses both clinical and histopathological aspects of the entity.
ASSOCIATED HISTOPATHOLOGIC ENTITIES OF IDIOPATHIC NONCIRRHOTIC PORTAL HYPERTENSION
NRH of the liver was first described by Ranstrom in 1953 [25], in a patient with Felty’s syndrome, and was called “miliary hepatocellular adenomatosis.” The term NRH was used by Steiner in 1959 [26] as a descriptive histopathologic term to be distinguished from cirrhosis. Subsequently, case reports and case series have revealed that a subset of patients with NRH present with portal hypertension [7]. This association was confirmed in a large-scale autopsy study, wherein 2.6% of cases had NRH, and 4.7% of these were found to have evidence of portal hypertension. Moreover, all NRH cases showed obliterative changes of the portal veins [27].
ISC was described by Popper in 1966 [28] as a subtype of macronodular cirrhosis, wherein inconspicuous, large regenerative nodules are vaguely delineated by thin and frequently incomplete septa. Some authors postulated that ISC represents regressed cirrhosis [29].
PNT of the liver was described by Sherlock et al. in 1966 [30], in four cases of portal hypertension, three of which were from autopsy. In PNT, the liver parenchyma adjacent to the hilum shows macroscopic and microscopic nodular transformation without advanced fibrosis, while the periphery is either atrophic or normal. The authors coined the term PNT to avoid confusion with other nodular lesions of the liver, such as cirrhosis (diffuse nodular transformation with extensive fibrosis), focal nodular hyperplasia (focal nodularity usually in the periphery, without portal hypertension), and NRH (diffuse nodular transformation without significant fibrosis). Histologic features of OPV were not evaluated in this original study; however, subsequent morphometric study suggested that portal vein obliteration might be involved in the pathogenesis of PNT [31].
Although these entities were initially reported as distinct disorders, subsequent morphologic studies demonstrated that NRH, ISC, and PNT had overlapping clinical and pathologic features with INCPH, supporting that these histopathologic entities share a common etiopathogenesis and most likely represent part of the histologic spectrum of a single condition [4-7].
EPIDEMIOLOGY
INCPH is commonly reported in developing countries and in lower socioeconomic groups [32,33]. INCPH is a common cause of portal hypertension in Japan and the Indian Subcontinent, constituting up to 30% and 40% of the cases, respectively, while only 3%–5% of portal hypertension in Western countries is attributed to INCPH [32,34,35]. However, the true prevalence of INCPH might be higher since patients are frequently misdiagnosed as having cirrhosis. Additionally, a significant proportion of such patients is in the subclinical phase of INCPH and might go unrecognized [3,12,27].
A male predilection has been reported in India and the West, whereas INCPH is more common in women in Japan [36-40]. The age of onset of INCPH tends to be younger in patients from India (25–35 years) compared to Japan (43–56 years) [36-40]. Limited data from the West have shown that the median age of onset is about 40 years [8]. INCPH has also been reported in children [41-45].
ETIOLOGY
No definite etiology is identified in more than half of the patients with INCPH [46]. Nevertheless, INCPH has been frequently reported in a multitude of immunologic disorders, including systemic lupus erythematosus, myasthenia gravis, systemic sclerosis, celiac disease, thyroiditis, rheumatoid arthritis, Crohn’s disease, Felty’s syndrome, Sjogren’s syndrome, autoimmune hepatitis, primary biliary cirrhosis, common variable immunodeficiency, and hypergammaglobulinemia, raising the possibility of an immunologic cause as an underlying etiology [8,25,46-55]. A survey from Japan showed that about 70% of female patients with INCPH had anti-DNA antibodies, and 24% and 21.5% showed antinuclear antibodies and antimicrosomal antibodies, respectively [2,56]. Likewise, an increased incidence of immune complex-associated glomerulonephritis was reported in INCPH patients following spleno-renal shunt, compared to those with normal liver [57].
Higher prevalence of INCPH in lower socioeconomic groups and experimental animal studies indicate an infectious etiology [3,52,58,59]. Especially in the West, INCPH is increasingly recognized in human immunodeficiency virus (HIV) patients [3,32]. Earlier studies have postulated that the use of didanosine, an antiviral medication of the reverse transcriptase inhibitor class with a potential for mitochondrial toxicity, might be associated with development of INCPH [60,61]. However, a recent multicenter case-control study showed that some of these patients were genetically predisposed to develop this condition [62]. In that study, a subset of HIV patients with prior exposure to didanosine and who subsequently developed INCPH was found to be associated with higher frequency of four specific single-nucleotide polymorphisms (SNPs) at the two genes coding enzymes of purine metabolism [62]. Moreover, the cumulative risk of developing INCPH was postulated to be 100% in the presence of all four SNPs. Alternatively, direct virus-induced sinusoidal endothelial cell injury might lead to INCPH in HIV patients [63]. Therefore, it is difficult to identify the precise cause of INCPH in HIV patients.
In addition to didanosine in HIV patients, exposure to various medications, chemicals, and toxins has been reported to be associated with INCPH [3,8,32,36]. For example, history of pica was noted in 46% of INCPH patients in Iran [64], and radiation and chemotherapy have been reported to result in INCPH [65,66].
Occurrence of INCPH in patients with congenital disorders including Adams-Oliver syndrome, Turner syndrome, phosphomannose isomerase deficiency, and familial cases of INCPH indicate a certain genetic makeup in these patients [3,45,67-72], making them susceptible to INCPH. In a report of four families with INCPH, six of seven members (85.5%) with INCPH were shown to be HLA-DR3 positive [72].
Lastly, the association between hypercoagulability and INCPH is relatively well established. Up to 54% of INCPH patients have been reported to be thrombophilic [6,32,73], with secondary portal vein thrombosis being relatively common. In addition, some of the characteristic histologic features of INCPH, such as obliteration and muscular hypertrophy of portal venous branches, might be explained by a prior/persistent thromboembolic event [3]. Thrombotic changes have also been noted within the portal veins and their larger branches in autopsies of INCPH patients [4]. In HIV patients, acquired protein S deficiency with resultant thrombophilia and INCPH has been reported [74], suggesting a role of hypercoagulability in these patients. Likewise, liver biopsies from patients with primary portal vein thrombosis without cirrhosis frequently show phlebosclerosis and NRH [66,75]. Given the association between INCPH and hypercoagulability, anticoagulation has been advocated as a potential treatment option for INCPH [6].
CLINICAL PRESENTATION
Patients with INCPH usually present with signs and symptoms associated with complications of portal hypertension, including upper gastrointestinal variceal bleeding, splenomegaly, and hypersplenism (anemia, thrombocytopenia, and leukopenia). The hepatic venous pressure gradient, the difference between wedged and free hepatic venous pressures, is significantly lower in INCPH than in cirrhosis and might be normal or only mildly elevated. In contrast, portal venous pressure is markedly elevated. These findings are indicative of presinusoidal portal hypertension [32,58,76]. Anorectal varices are also common in INCPH, but bleeding from anorectal varices is uncommon [77]. Ascites, encephalopathy, hepatorenal syndrome, and jaundice can occur with a lower frequency, and some patients present with extrahepatic portal vein thrombosis [3,12,32,78]. Liver enzymes can be normal or slightly abnormal; presentation with isolated liver enzyme abnormalities was previously reported in 20% of INCPH cases [46]. Hepatic synthetic function is mostly preserved but can be rarely compromised, requiring liver transplantation [79].
IMAGING STUDIES
Ultrasonography might show nodularity of the liver surface and thickened portal venous wall [4,78,80]. Computed tomography and magnetic resonance imaging can show signs of portal hypertension, extrahepatic portal vein thrombosis, intrahepatic portal abnormalities, nodular liver contour, and hypertrophy of the caudate lobe with atrophy of segment IV. The latter two features are more commonly seen in cirrhosis [70,81]. When measured using elastography, the mean liver stiffness in INCPH (8.4±3.3 kPa by transient elastography and 1.56 [0.98–2.37] m/sec by acoustic radiation force impulse elastography) is lower than that of cirrhosis (40.9±20.5 kPa and 2.44 [1.08–3.83] m/sec, respectively) [76,82].
PATHOLOGIC FEATURES
The gross appearance of the liver is heterogeneous and can be normal, enlarged, or atrophic with a smooth, wrinkled, or nodular surface (Fig. 1) [9,21,32]. In patients requiring liver transplantation for advanced INCPH, the explanted liver tends to be atrophic with frequent surface nodularity [12,79]. Subcapsular septation, prominence of portal tracts near the surface, and sclerosis of portal vein branches with or without organized thrombi have been described [9,21,40,83]. Relative hypertrophy of the right lobe and atrophy of the left lobe are common findings [21]. The cut surface might be slightly nodular or partially nodular near the hepatic hilum [21,79].
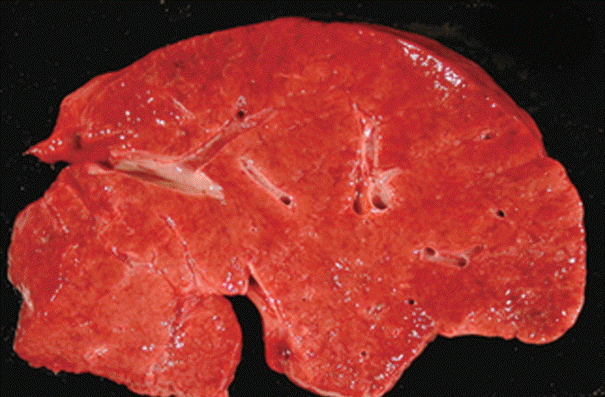
Gross cut surface of a liver with idiopathic noncirrhotic portal hypertension demonstrates vague nodularity without cirrhosis.
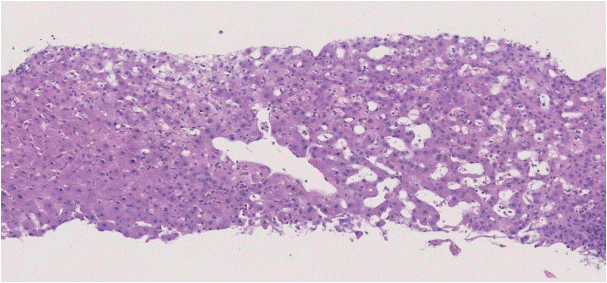
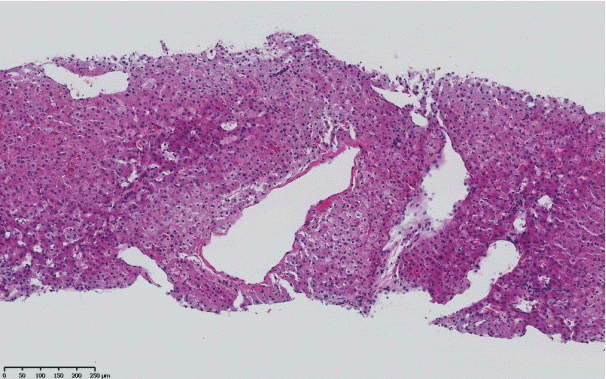
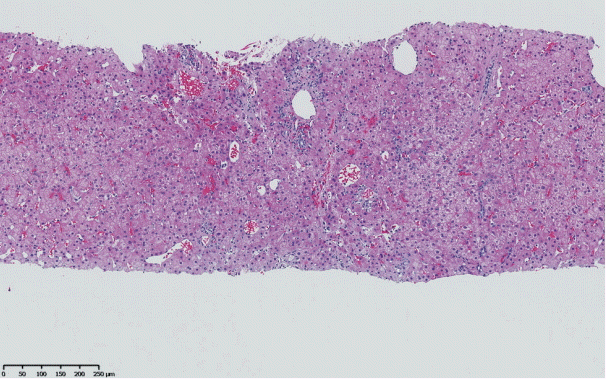
The histopathological features of INCPH vary and are dependent on the phase of the disease as well as the area sampled [40]. Variable combinations of these histological components such as OPV, variable portal fibrosis, vascular abnormalities, and NRH can be seen. The hallmark of INCPH is OPV, characterized by dense fibrosis/sclerosis of the portal vein along with portal/periportal fibrosis, phlebosclerosis of portal vein branches with resultant decrease of the lumen (Fig. 2), an increase in the number of portal vascular channels (Fig. 3), and arterialization of the portal vein branches. In addition, there can be portal shunting vessels that directly connect periportal areas with the hepatic lobule (Fig. 4). Mild lymphocytic portal inflammation and mild bile ductular proliferation might be seen [11,12,21,66]. Changes in the lobules include diffuse or focal nodular regeneration (Fig. 5), dilated sinusoids (megasinusoids) (Fig. 6), increased number of venous profiles per lobule (Fig. 7), with architectural distortion (Fig. 8), and incomplete septa, i.e., slender fibrous septa originating from a portal tract that blindly ends in the lobule, perisinusoidal fibrosis, and perivenular fibrosis [3,6,8,11,12,32,66,79]. Portal tract remnants, or rudimentary/hypoplastic portal tracts—small portal tracts wherein the lumen of the bile duct or artery is smaller than adjacent hepatocytes, with inconspicuous or sometimes absent portal vein branches—might be identified (Fig. 9). The above histologic features are consistently reported in INCPH; however, the specificity of individual histologic findings remains unclear. For example, portal fibrosis and portal venous obliteration have also been seen in control livers without INCPH [4]. Similarly, the histologic features of INCPH were seen in patients with multiple comorbidities, without established diagnosis of INCPH [11].
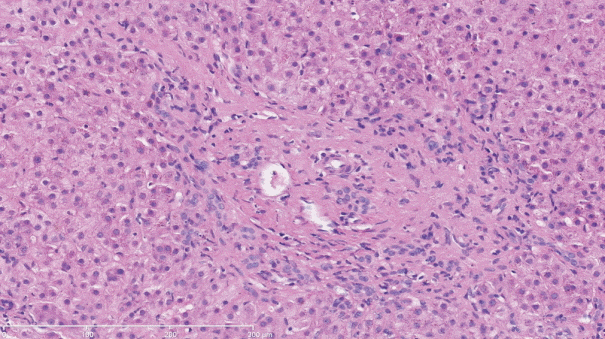
Phlebosclerosis with narrowed venous lumen.
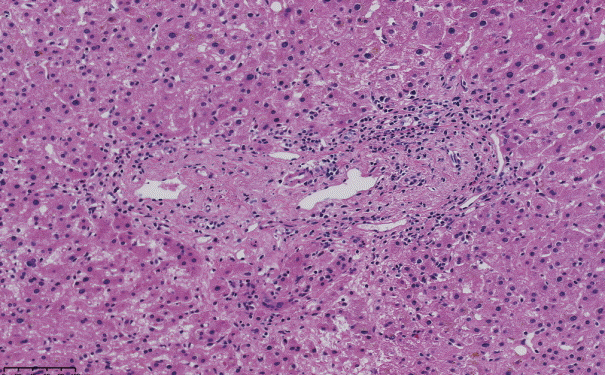
Portal sclerosis associated with increased number of vascular channels.
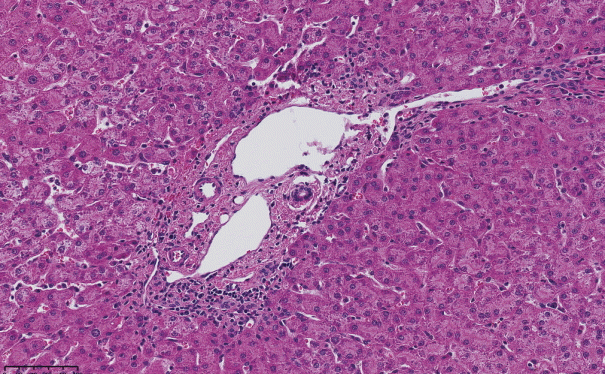
Abnormally dilated portal venous branches associated with herniation of the vein into the hepatic lobule (shunt vessel).
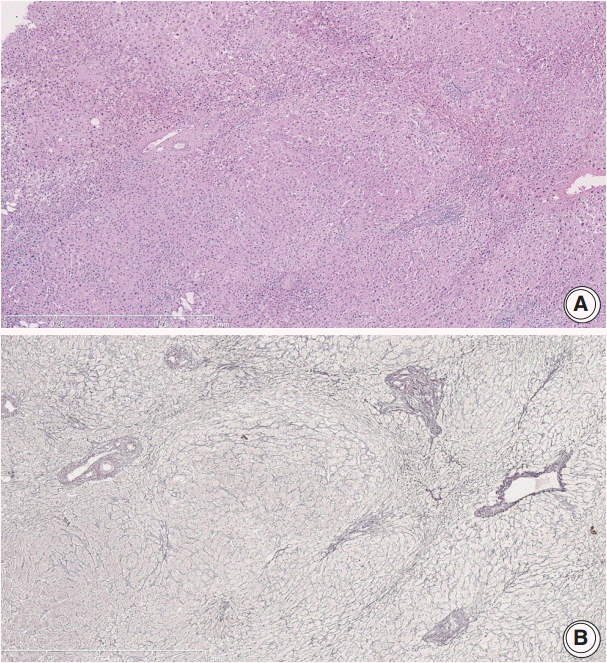
(A) Regenerative nodules of the lobule without cirrhosis. (B) Reticulin stain of the corresponding area highlights regenerative nodules.
Dilated sinusoids (also known as megasinusoids).
Abnormally dilated veins in the lobule.
Distorted hepatic architecture in idiopathic noncirrhotic portal hypertension.
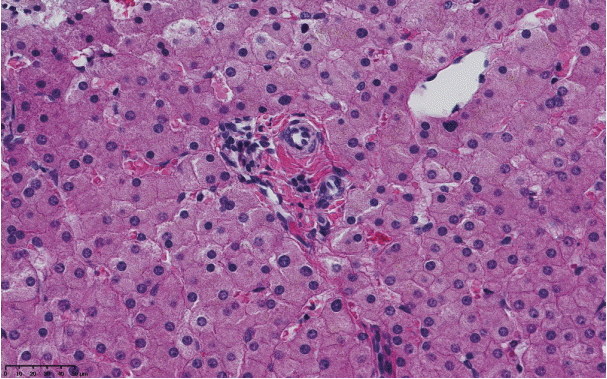
Hypoplastic/rudimentary portal tract.
Verheij et al. [66] evaluated variable histologic features of INCPH in Western patients and found that portal tract remnants, phlebosclerosis of portal vein branches, and NRH are more common in INCPH compared to noncirrhotic portal vein thrombosis. In contrast, sinusoidal dilatation, shunting vessels, increased number of portal vessels, and increased number of veins per lobule were suggested to represent secondary changes of portal hypertension [66]. In addition, NRH was significantly more common in HIV-associated INCPH, whereas portal tract remnants were frequent in INCPH without HIV. Nevertheless, no correlation has been found between the histomorphology and clinical signs of portal hypertension [66].
TREATMENT AND OUTCOME
The treatment primarily consists of controlling and preventing the symptoms of portal hypertension, especially variceal bleeding. The management strategy used for cirrhotic patients with portal hypertension is currently being used for INCPH, with a favorable long-term outcome [10]. For example, acute hemorrhage from esophageal varices is treated with combined vasoactive drugs and endoscopic variceal ligation/sclerotherapy. Transjugular intrahepatic porto systemic shunting (TIPS) can be offered to patients who fail to respond to endoscopic therapy or those with recurrent bleeding. Prophylaxis for variceal bleeding consists of the use of non-selective beta blockers, endoscopic variceal ligation, or TIPS in selected patients [3,84]. In addition, any drugs associated with development of INCPH are discontinued, and medical conditions associated with INCPH should be treated [3].
Preliminary data suggests that anticoagulation for thrombophilic INCPH patients might be beneficial. In a case series, eight of 15 INCPH patients with complete or partial portal vein thrombosis responded to anticoagulation with some degree of recanalization [10]. Also, early anticoagulation for INCPH patients with hypercoagulability appeared to show a favorable clinical outcome (no death or liver transplantation) [46], and anticoagulation therapy improved liver function tests in a patient with HIV and INCPH [85].
A small number of INCPH patients have undergone liver transplantation for complicated portal hypertension. Most of these patients carried a presumed diagnosis of cirrhosis prior to transplant [12,79]. Two patients developed histologic features of INCPH in the allograft biopsies within 1 year of the transplantation, and one of them subsequently developed recurrent portal hypertension [12].
Although variceal bleeding is common, the overall long-term prognosis of INCPH appears to be better than that of cirrhosis, possibly due to preserved hepatic function in a majority of the patients [3,10,20,46].
CONCLUSION
INCPH is a rare condition that has been under-recognized both clinically and pathologically. Many different terms have been used to describe this entity, adding to the confusion. Although its management primarily focuses on the control and prophylaxis of complications of portal hypertension, the etiopathogenesis and natural history of INCPH appear distinct from those of cirrhosis. Recognition of the clinical presentation, histopathology, and associated risk factors of INCPH will enable the correct classification of patients with INCPH.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.