Ménétrier’s Disease: Its Mimickers and Pathogenesis
Article information
Abstract
Ménétrier’s disease is a rare protein-losing hypertrophic gastropathy. Histologically, it can be mistaken for other disorders showing hypertrophic gastropathy. The pathogenesis of Ménétrier’s disease is not fully understood; however, it appears that the epidermal growth factor receptor (EGFR) ligand, transforming growth factor alpha, contributes to the pathogenesis of this disorder. In this review, we will discuss disease entities that can mimic Ménétrier’s disease and the role of EGFR signaling in Ménétrier’s disease.
Ménétrier’s disease (MD), hypoproteinemic hypertrophic gastropathy, is a rare acquired disorder characterized by giant gastric rugal folds in the body and fundus, often with antral sparing, decreased acid secretion, increased gastric mucus production, and hypoalbuminemia secondary to protein loss in the gastric mucosa [1]. It affects men more frequently than women, and the typical age at diagnosis is between 30 to 60 years.
CLINICAL PRESENTATION
Typical clinical presentations include nausea, vomiting, diarrhea, abdominal pain, weight loss, malnutrition, and peripheral edema due to hypoalbuminemia. MD usually shows an insidious onset with progressive features and is associated with an increased risk of gastric cancer [2,3]. Variants with abrupt onset and spontaneous remission have been reported. These variants are associated with cytomegalovirus (CMV) or Helicobacter pylori infection [4-6]. CMV-associated cases usually occur in children; however, cases in adults have also been reported [7-10]. MD has also been reported in patients with autoimmune diseases such as inflammatory bowel disease, sclerosing cholangitis, and ankylosing spondylitis, suggesting an immunological component to its pathogenesis [11,12]. Endoscopically, the markedly thickened gastric mucosal folds resemble cerebral convolutions and primarily affect the body and fundus but spare the antrum (Fig. 1). Gastric pH is increased due to loss of parietal cells, and copious thick mucus production is seen secondary to foveolar hyperplasia [2,3].
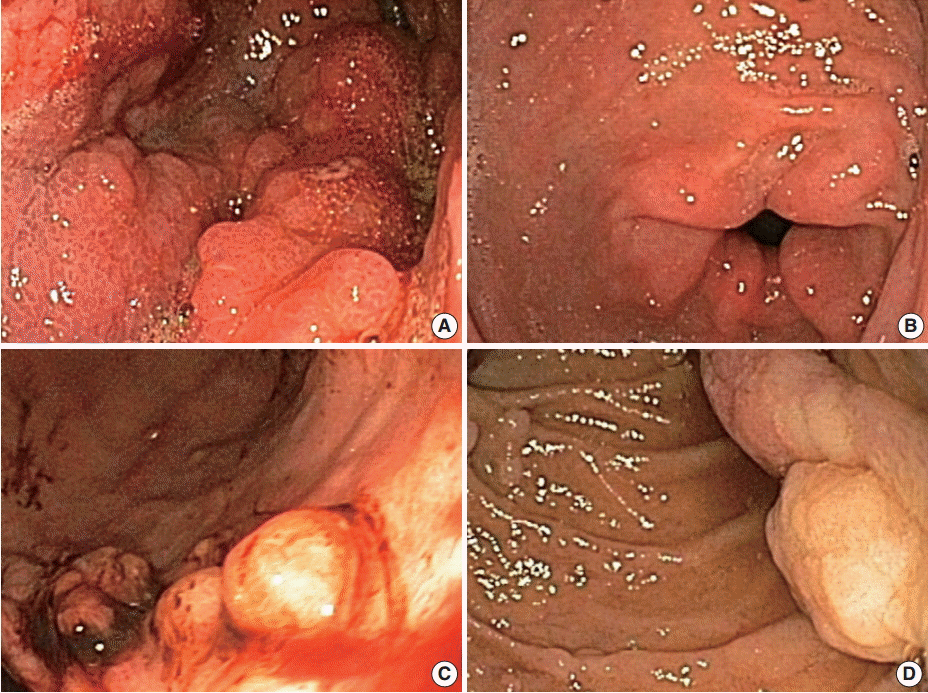
Endoscopic appearance of Ménétrier’s disease and juvenile polyposis syndrome. (A) Gastric body of Ménétrier’s disease patient with diffuse hypertrophic gastric folds. (B) Gastric antrum of the same patient is not involved. (C) Gastric body of juvenile polyposis syndrome patient with multiple sessile polyps. (D) Juvenile polyps in the duodenum.
MICROSCOPIC FEATURES
Foveolar hyperplasia that often results in mucosal thickness 1 cm or greater is the most striking feature of MD. The foveolar epithelium shows a corkscrew morphology due to massive foveolar hyperplasia, but the overall linear architecture is maintained (Fig. 2A). Oxyntic glands are atrophic with reduced or absent parietal cells, and deep glands can be cystically dilated. The lamina propria shows a variable amount of predominantly chronic inflammatory cell infiltration with scattered eosinophils. Prominent vertical strands of smooth muscle in the lamina propria are also identifiable (Fig. 2D).
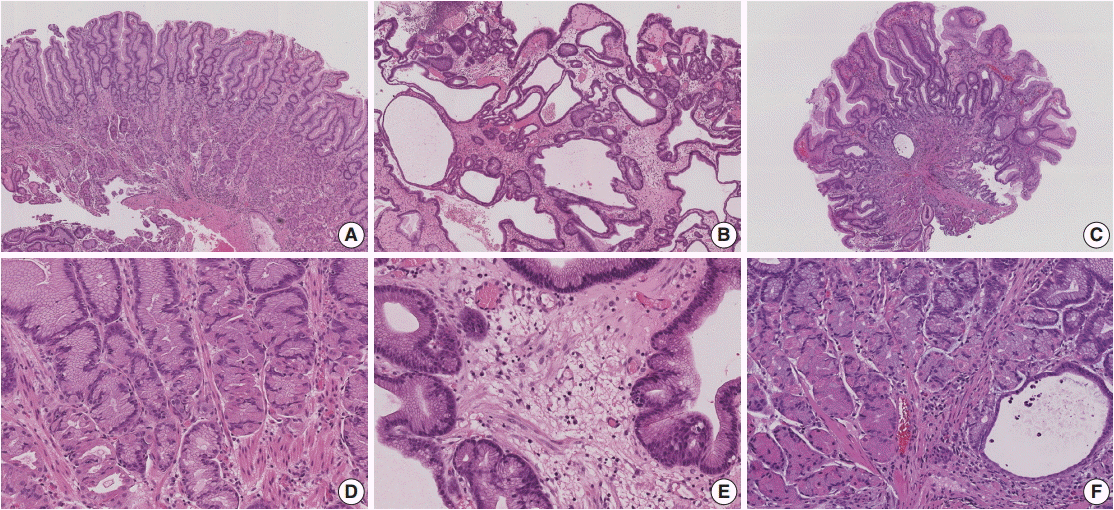
Histologic comparison of Ménétrier’s disease and gastric polyps. (A) Ménétrier’s disease shows foveolar hyperplasia with corkscrew morphology and cystically dilated deep glands; however, overall linear architecture is maintained. (B) Juvenile polyp shows foveolar hyperplasia with cystically dilated superficial and deep glands. Linear architecture is disrupted. (C) Peutz-Jeghers polyp shows foveolar hyperplasia and cystically dilated glands. Lamina propria of Ménétrier’s disease shows strands of smooth muscle bundles (D) whereas lamina propria of juvenile polyp is edematous without prominent smooth muscle (E). (F) Peutz-Jeghers polyp shows arborizing smooth muscle strands in the lamina propria, which is less prominent than counterparts in small intestine or colon.
DIFFERENTIAL DIAGNOSIS
Diseases that show thickened gastric folds can mimic MD at endoscopy. These conditions range from foveolar hyperplasia in reactive conditions to malignancy (Table 1). Because some of these diseases share histologic characteristics with MD, correlation of microscopic findings with endoscopic and clinical features is important in order to establish the correct diagnosis.

Differential diagnosis of Ménétrier’s disease
Because accurate diagnosis of some of these diseases requires examination of very thick gastric mucosa, large snare biopsies that capture the entire thickness of the mucosa are recommended instead of standard forceps biopsies. Disease entities with diffuse hypertrophic gastropathy include hypertrophic lymphocytic gastritis, hypertrophic hypersecretory gastropathy, and Zollinger-Ellison syndrome (ZES). Hypertrophic lymphocytic gastritis often presents as giant fundic mucosal folds with relative sparing of the antrum similar to MD. In contrast to MD, the gastric mucosa in this condition shows diffuse and severe inflammation with prominent intraepithelial lymphocytes. Foveolar hyperplasia is confined to areas with inflammation [13]. Hypertrophic hypersecretory gastropathy is a rare acquired gastropathy that involves hypersecretion of acid, pepsin, and mucin. Endoscopically, it is characterized by hypertrophic gastric folds and “cobblestone” gastric body mucosa with atrophic antral mucosa. Histologically, it is differentiated from MD in that hyperplasia is seen in both the foveolar epithelium and oxyntic glands. Gastric glands with cystic dilatation can also be seen [14,15]. ZES is characterized by ectopic gastrin secretion (gastrinoma), increased gastric acid secretion, and intractable peptic ulcer disease, and it shows diffusely thickened gastric folds, especially in the body and fundus. Multiple peptic ulcers can also be observed. Histologic characteristics of ZES include diffuse parietal cell hyperplasia and hypertrophy. Parietal cells extend to the base of the glands and into the antrum. However, the foveolar epithelium does not show hyperplasia. Nodular and linear enterochromaffin-like cell hyperplasia can frequently be observed in the disease and is associated with multiple endocrine neoplasia type 1.
Gastric polyps can manifest as focal hypertrophic gastropathy and can be numerous and diffusely distributed in polyposis syndromes, resulting in a hypertrophic appearance. Gastric hyperplastic polyps are the most common, accounting for 70% of gastric epithelial polyps [16]. The antrum is the most common site for hyperplastic polyps; however, they can develop in other areas of the stomach and can be solitary or multiple. Hyperplastic polyps usually develop in the background of other gastric pathology such as atrophic gastritis, reactive gastropathy, or any form of acute or chronic gastritis. The polyps show foveolar hyperplasia with dilated and tortuous glands. Foci of intestinal metaplasia can be seen in 15% of lesions. Surface erosions and ulcerations are frequently observed and associated with reactive changes.
Juvenile polyposis syndrome (JPS) is characterized by hamartomatous polyps in the gastrointestinal tract, most commonly in the colon. Most colonic juvenile polyps are sporadic, but germline mutations in either BMPR1A or SMAD4 genes have been detected in 50% of cases of JPS, which is inherited in an autosomal dominant pattern. A severe gastric phenotype has been shown in SMAD4 mutations, particularly nonsense mutations in exon 11 and a deletion of four base pairs in exon 9 at codon 415 [17]. Histologically, gastric polyps in JPS are similar to other hyperplastic or hamartomatous polyps and are characterized by foveolar hyperplasia. Compared to MD, gastric juvenile polyps show distorted glandular architecture, edematous stroma, lower gland to stroma ratio, and less conspicuous eosinophils and smooth muscle fibers in the lamina propria (Table 2, Fig. 2B, E). Among 48 patients referred to Vanderbilt University Medical Center with a diagnosis of MD for a clinical trial of cetuximab conducted in individuals with advanced MD, 25 (52%) were confirmed with the diagnosis of MD. The most common entities mistaken for MD were gastric hyperplastic polyps or JPS, found in seven of the total 23 non-MD patients (30%) [8].
Microscopic features of Ménétrier’s disease, hyperplastic polyp, and juvenile polyp in the gastric body/fundus
Gastric polyps in syndromes with both gastrointestinal and extra-intestinal manifestations can also mimic MD; therefore, clinical information is essential for a correct diagnosis. Cronkhite-Canada syndrome (CCS) is a rare nonhereditary syndrome characterized by diffuse polyposis in the gastrointestinal tract, alopecia, cutaneous hyperpigmentation, and onychodystrophy [18]. Gastric polyps of CCS share many histologic features with juvenile polyps. Mixed inflammation in the lamina propria with prominent eosinophils and eosinophilic infiltration of glandular epithelium with crypt abscesses is a helpful upper gastrointestinal feature of CCS. Peutz-Jeghers syndrome (PJS) is characterized by hamartomatous gastrointestinal polyps with increased skin and mucosal pigmentation. PJS is associated with mutations in the serine/threonine kinase 11 (STK11) gene and shows an autosomal dominant inheritance pattern. PJS polyps are usually characterized by hyperplastic epithelium divided by arborizing bands of smooth muscle. However, PJS polyps in the stomach show dilated hyperplastic glands and less prominent smooth muscle strands compared to PJS polyps in other gastrointestinal tract (Fig. 2C, F) [19]. This makes it challenging to differentiate them from hyperplastic polyps or MD. Cowden syndrome is characterized by numerous hamartomas in the gastrointestinal tract and extraintestinal sites such as skin, breast, thyroid, gynecologic, oral cavity, and central nervous system. Mutations in the phosphatase and tensin homolog (PTEN) gene are detected in up to 80% of Cowden syndrome cases and show an autosomal dominant inheritance pattern [20]. A diffuse “carpetlike” distribution of white mucosal plaques in the esophagus is a characteristic endoscopic finding, which microscopically shows thickened squamous epithelium with glycogen accumulation in the cytoplasm (glycogenic acanthosis) [21]. Histologic features of Cowden syndrome overlap those of hyperplastic polyps or juvenile polyps.
Gastric adenocarcinoma and proximal polyposis of the stomach is a recently described entity with an autosomal dominant inheritance with incomplete penetrance. The genetic cause of this disorder is unknown. Numerous gastric polyps carpeting the proximal stomach are observed endoscopically with sparing of the esophagus, antrum, duodenum, and colon. These polyps histologically overlap with fundic gland polyps with multiple foci of low- and high-grade dysplasia. Occasional hyperplastic or adenomatous polyps can occur. This syndrome is associated with a high risk of intestinal-type gastric adenocarcinoma, and 41 cases in five families have been reported to date. Nine of the cases developed gastric adenocarcinoma [22,23]. Other entities that can mimic hypertrophic gastropathy include diffuse gastric carcinoma, gastric lymphoma, and amyloidosis.
TRANSFORMING GROWTH FACTOR ALPHA IN MÉNÉTRIER’S DISEASE
The pathophysiology of MD has not been fully elucidated. However, the observation that transgenic mice overexpressing transforming growth factor alpha (TGF-α) in the stomach develop gastric changes that resemble MD and increased TGF-α expression in the gastric mucosa of MD patients suggest an important role of this growth factor in its pathogenesis [24-31].
TGF-α is one of seven mammalian ligands that bind epidermal growth factor receptor (EGFR) [32]. EGFR is a receptor tyrosine kinase that mediates signal transduction in many cell types. A common action of TGF-α is increased cellular proliferation, and elevated TGF-α expression has been associated with neoplastic transformation [33,34]. Transgenic mouse models that overexpress TGF-α showed epithelial hyperplasia of liver, pancreas, stomach, small intestine, colon, mammary glands, and coagulation glands. Acinoductular metaplasia was also found in the pancreas, and neoplasia was identified in the mammary glands and liver [25,28,31]. Interestingly, the stomachs of mice that overexpressed TGF-α showed characteristic MD features, such as foveolar hyperplasia, glandular cystic dilatation, oxyntic atrophy, increased mucin production, and reduced acid secretion [24,26,27]. Moreover, it has been shown that TGF-α expression in both RNA and protein is increased in the gastric mucosa of MD patients [24,29,30]. Functionally, TGF-α overexpression appears to direct gastric stem/progenitor cells to surface mucous cell differentiation at the expense of parietal and chief cell differentiation [35-37]. Additionally, TGF-α overexpression repatterns gastric fundic-type epithelium into antral-type epithelium, as evidenced by aberrant expression of the gastric antral markers, Pdx1 and gastrin, in the gastric body of both TGF-α transgenic mice and MD patients [38].
Involvement of EGFR signaling in the pathogenesis of MD is further supported by a CMV-associated variant of MD. It has been shown that the CMV principal envelope glycoprotein, gB, binds EGFR, induces EGFR-HER3 hetero-oligomers, and activates EGFR signaling [39]. In addition, disruption of the bone morphogenetic protein/TGF-β signaling pathway in mice can lead to increased levels of TGF-α and amphiregulin, another EGFR ligand [40-42]. We also found that phosphorylated EGFR is increased in the involved gastric mucosa of JPS patients (Fig. 3). These findings could contribute to the histolopathologic similarities between gastric JPS and MD.
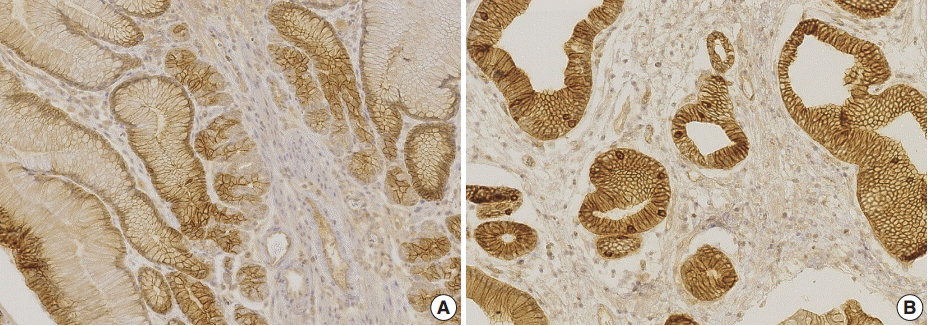
Immunohistochemistry of phosphorylated EGFR (pEGFR). Hyperplastic foveolar epithelium of both Ménétrier’s disease (A) and juvenile polyp (B) shows membranous pEGFR staining.
TREATMENT
It is advised to rule out spontaneous remission variants associated with CMV or Helicobacter pylori infection by appropriate clinical testing. CMV-associated MD usually resolves within several weeks to months [4,43]. H. pylori eradication can be attempted if the organism is detected. Other than these approaches, there is currently no standard medical treatment for MD. Reports on efficacy of steroids, anticholinergics, acid suppression, and octreotide are inconsistent [8]. Partial or total gastrectomy is reserved for patients with intractable or debilitating disease and for cases with a high risk of developing gastric cancer. The link to TGF-α and EGFR signaling in the pathogenesis of MD led us to design a clinical trial using a neutralizing monoclonal antibody (cetuximab) against EGFR for MD patients. All seven patients who completed a month of cetuximab treatment showed statistically significant improvement in quality-of-life indices, increased parietal cell mass and gastric acidity. In addition, four patients showed near-complete histological resolution. To our surprise, improvement in symptoms usually started within one to two days after the first cetuximab infusion, and parietal cell mass was significantly increased within 24 hours [11]. Therefore, based on the results of this study, cetuximab should be considered a therapeutic option for MD patients.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was supported by National Cancer Institute R01 CA46413 and P50 CA95103 Specialized Programs of Research Excellence grants to RJC.