Epstein-Barr Virus–Associated Lymphoproliferative Disorders: Review and Update on 2016 WHO Classification
Article information
Abstract
Epstein-Barr virus (human herpesvirus-4) is very common virus that can be detected in more than 95% of the human population. Most people are asymptomatic and live their entire lives in a chronically infected state (IgG positive). However, in some populations, the Epstein-Barr virus (EBV) has been involved in the occurrence of a wide range of B-cell lymphoproliferative disorders (LPDs), including Burkitt lymphoma, classic Hodgkin’s lymphoma, and immune–deficiency associated LPDs (post-transplant and human immunodeficiency virus–associated LPDs). T-cell LPDs have been reported to be associated with EBV with a subset of peripheral T-cell lymphomas, angioimmunoblastic T-cell lymphomas, extranodal nasal natural killer/T-cell lymphomas, and other rare histotypes. This article reviews the current evidence covering EBV-associated LPDs based on the 2016 classification of the World Health Organization. These LPD entities often pose diagnostic challenges, both clinically and pathologically, so it is important to understand their unique pathophysiology for correct diagnoses and optimal management.
Epstein-Barr virus (EBV) is classified as a γ-herpes virus and contains a linear DNA molecule about 172 kb in length, which affects more than 90% of the worldwide adult population. If the infection does not become clinically silent, infectious mononucleosis is experienced by the exposed persons [1]. Although EBV infection is lifelong, a long latency and reactivation of EBV results in various lymphoproliferative lesions including hematologic malignancies [2]. This article reviews the current understanding of EBV-associated lymphoproliferative disorders (LPDs) based on the 2016 classification of the World Health Organization (WHO) [3]. Some LPDs exhibit a predisposition in Asian populations, including Koreans. These entities often pose diagnostic challenges, both clinically and pathologically, and it is important to understand their unique pathophysiology for correct diagnoses and optimal management. Generally intrinsic defects and post-transplant lymphoproliferative disorders (PTLDs) are excluded from this review, these are treated separately as immunodeficiency disorders by the WHO.
EPSTEIN-BARR VIRUS–ASSOCIATED B-CELL LYMPHOPROLIFERATIVE DISORDERS
The spectrum of EBV-associated B-cell LPDs is broad, ranging from reactive lymphoproliferative lymphadenitis to lymphomas. All the related disease entities are shown in Table 1. Well-defined lymphoma entities such as Burkitt lymphoma, classical Hodgkin lymphoma, and plasmablastic lymphoma are not included [4].
Epstein-Barr virus.associated B-cell lymphoproliferative diseases
Infectious mononucleosis
Primary EBV infection occurs most often in childhood and is generally asymptomatic. In adolescence, it is associated with a self-limiting infectious mononucleosis syndrome, manifested by fever, pharyngitis, malaise, and atypical lymphocytosis. Following primary infection, most individuals remain a life-long carrier of the virus without serious sequelae [5]. However, a small population with the latent infection will develop various LPDs.
EBV is transmitted from the host by saliva and infected EBV replicates within oropharyngeal epithelium and is then exposed to circulating B-lymphocytes. Peripheral EBV-infected memory B-cells can return to the Waldeyer’s ring, undergoing reactivation to produce an infectious virus that will be shed in the saliva. EBV-specific cytotoxic T-cells (CTL) destroy most infected cells.
The histologic features of infectious mononucleosis vary during the course of the disease. Early in the disorder, follicular hyperplasia occurs with monocytoid B-cell aggregates and epithelioid histiocytes. Later, the expansion of the paracortex predominates. The immunoblasts resemble classical Reed-Sternberg (RS) cells. The immunoblasts of predominantly B-cell types and partly T-cell types often express CD30. In-situ hybridization (ISH) of EBV-encoded small RNAs (EBERs) exhibit numerous positive immunoblasts in the paracortex but not in the germinal centers [6].
Chronic active EBV of B-cell types
As first defined by Lekstrom-Himes et al. [7], chronic active EBV (CAEBV) of B-cell types refers to a chronic or persistent EBV infection characterized by a severe illness lasting more than 6 months, persistent elevated EBV titers, and evidence of EBV-related organ damage. Currently, CAEBV is defined as (1) a severe progressive illness with a duration of more than 6 months, (2) lymphocytic infiltration of tissue (e.g., lymph nodes, lungs, liver, central nervous tissue, bone marrow, eye, and skin), (3) elevated EBV DNA and RNA in affected tissue, and (4) absence of any other immunosuppressive conditions [8].
Histologically, the lymph nodes exhibits features resembling polymorphic PTLD, with paracortical expansion, plasmacytoid lymphoblastic proliferation, presence of plasma cells, and presence of occasional RS-like cells. EBV-ISH positive B-cells are noted in the paracortex. Among CAEBV patients, 63% had clonal immunoglobulin rearrangement [9].
EBV-positive diffuse large B-cell lymphoma
EBV-positive diffuse large B-cell lymphoma (DLBCL), not otherwise specified was originally described as “senile EBV-associated B-cell LPD,” or “EBV-positive DLBCL of the elderly” (older than 50 years) (WHO 4th edition) [10]. However, subsequent studies have shown that EBV-positive DLBCL is not limited to this older age group. In the elderly group, it is thought to be related to immunosenescence, which modifies T-cell homeostasis through a lack of thymic output of naïve T-cells and an accumulation of viral specific CD8+ T cells.
Histologically, four types have been described: monomorphic (DLBCL-like, monotonous sheets of large cells), polymorphic in the inflammatory background, T-cell/histiocyte-rich large cell lymphoma, and plasmacytoid differentiation. Immunophenotypically, the tumor cells express pan B-cell markers (CD20, PAX5, CD79a, OCT-2, and BOB-1), and are mostly CD30+ , but lack CD15 expression.
This disease entity frequently involves extranodal sites including the skin, lung, tonsils, and stomach. Unfavorable prognostic factors include older age (>70 years), high international prognostic index, and activated B-cell phenotype [11].
EBV mucocutaneous ulcer
This self-limited EBV positive B-cell proliferation is characterized by the presence of mucocutaneous ulcers with an indolent clinical course. This phenomenon is likely due to a more localized form of decreased immune surveillance, which is supported by a very low EBV viral load. The frequently involved sites includes the skin, oropharyngeal mucosa, and gastrointestinal tract [12].
Histologically a sharply demarcated ulcer is lined by an inflammatory infiltrate with clusters of large atypical cells, often with RS cell-like features. Phenotypically the large atypical cells are variably positive for CD20 and CD30, and uniformly positive for EBV-ISH and CD15+ in half of the cases (Fig. 1).
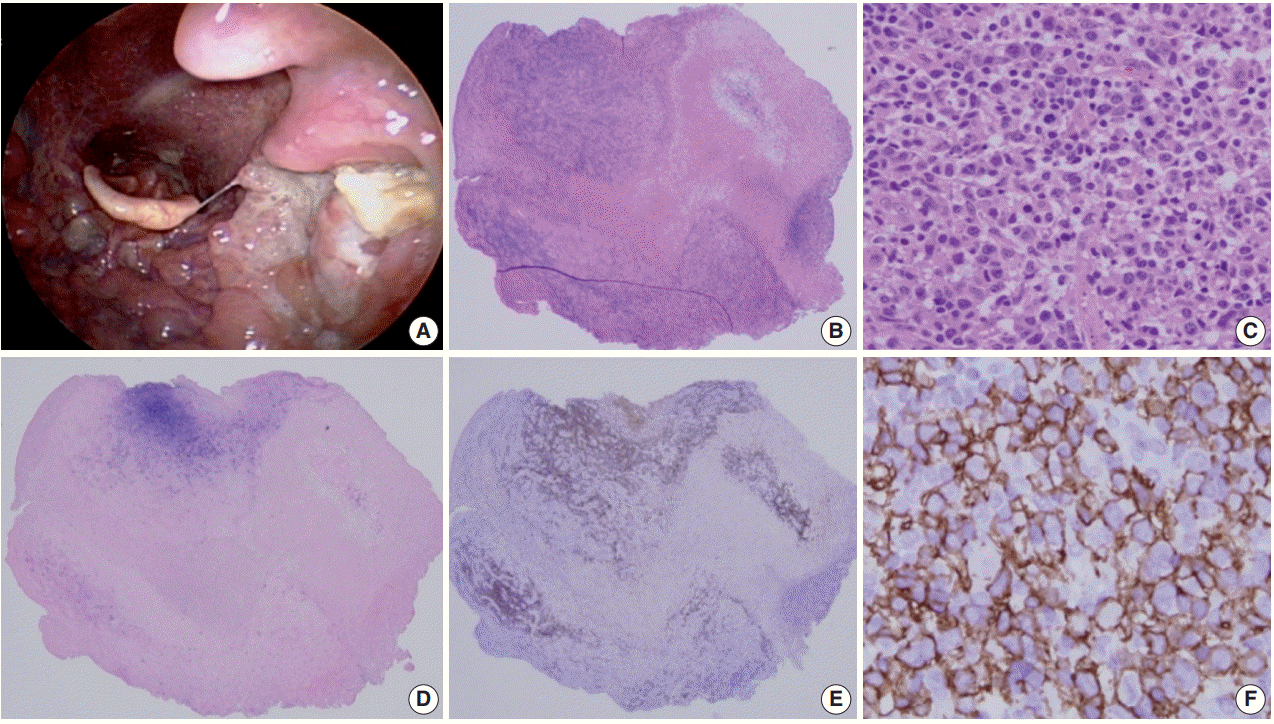
Mucocutaneous ulcer (Courtesy of Dr. J.H. Paik). (A) This 70-year-old female presented with a sore throat, painful swelling saliva, and tonsillar enlargement with a discrete ulcer. (B) The scanning power view shows a dense infiltrate beneath the ulcer. (C) Medium sized atypical lymphocytes are observed. (D) Epstein-Barr virus (EBV)–in-situ hybridization positive cells are aggregated in the ulcer bed. (E) CD20 immunostaining disclosed overlapping with EBV-positive cells. (F) The large atypical cells are diffusely and strongly positive for CD20.
DLBCL associated with chronic inflammation
DLBCL associated with chronic inflammation develops in the setting of long-standing chronic inflammation with EBV association. It usually involves body cavities or enclosed spaces (like cysts). Pyothorax-associated lymphoma (PAL) represents the prototype of this entity.
Cases with PAL have long history of chronic pyothorax and may present with chest pain, fever, cough, dyspnea, and tumor mass. The prognosis of PAL is poor.
The morphology discloses a diffuse proliferation of large atypical lymphocytes with plasmacytoid cytomorphology. Immunohistochemistry reveals neoplastic cells that represent pan-B cell markers, usually positive for IRF4/MUM1, and CD13. An aberrant expression of T-cell phenotypes is also seen.
The unique genomic instability has been reported as follows: A20 deletion, interferon-inducible 27 (IFI27), TP53 mutation, and MYC amplification [13].
Lymphomatoid granulomatosis
Katzenstein et al. [14] initially described a rare angiocentric and angiodestructive EBV-associated LPD, which was distinct from Wegener’s granulomatosis.
Nearly all patients present with symptoms related to pulmonary involvement, followed by involved sites of the central nervous system, skin, liver, and kidney. Radiologically, bilateral variable sized lung nodules are noted in lymphomatoid granulomatosis (LYG).
The histologic features of LYG are observed in lung nodules. All the lesions are angioinvasive and angiocentric with fibrinoid necrosis of the vascular wall. The infiltrate is polymorphous with an admixture of small lymphocytes, histiocytes, and large lymphoid cells. The grading of LYG is based on the proportion of EBV positive cells. In general, grade 1 and grade 2 lesions are approached using strategies that are designed to improve the host’s immune system, whereas grade 3 lesions require chemotherapy and do not respond to immunomodulatory therapies. Phenotypically, large atypical EBV-positive B cells express CD20, PAX5, CD79a, CD30+ , and CD15– [15].
EBV-ASSOCIATED T-CELL AND NATURAL KILLER CELL LPDS
EBV is a ubiquitous herpes virus with tropism for B cells, but the infection of T cells and natural killer (NK) cells may lead to several EBV-related LPDs. EBV-positive T/NK LPD encompasses disease entities with a broad clinicopathologic spectrum (Table 2) [16].
EBV-associated T-cell and NK cell lymphoproliferative diseases
EBV-associated hemophagocytic lymphohistiocytosis
Hemophagocytic lymphohistiocytosis (HLH) is a clinicopathologic syndrome encompassing a markedly dysregulated immune response and hypercytokinemia. HLH is characterized clinically by fever, splenomegaly, and cytopenias, and histologically by hemophagocytosis. The supportive laboratory findings for HLH are as follows: extremely high serum level of ferritin, lactate dehydrogenase, soluble CD25, and elevated viral capsid. EBV associated HLH accounts for 40% of HLH.
Histologically, EBV-positive T-cells and hemophagocytosing histiocytes are scattered in the sinusoids of the bone marrow and liver. The T-cells express CD8 and granzyme B. Even though it is uncommon, NK cells can be infiltrated in HLH [17].
HLH can be effectively controlled in most patients (more than 90%), but the other 10% often die of fulminant disease.
CAEBV infection of T-cell or NK-cell types, systemic
CAEBV infection was initially defined as follows: (1) markedly abnormal EBV antibody titer; (2) histologic evidence of organ involved- interstitial pneumonia, hypoplasia of the bone marrow, uveitis, lymphadenitis, persistent hepatitis, or splenomegaly; and (3) increased EBV RNA in affected tissue [18].
Clinically, CAEBV-T/NK is a disease of children but is also detected in young adults and even in middle-aged older adults with a mean age of 11.3 years [19]. The symptoms are usually prolonged fever, hepatomegaly, splenomegaly, thrombocytopenia, anemia, and lymphadenopathy [19]. Life-threatening complications include hemophagocytic syndrome, interstitial pneumonia, malignant lymphoma, coronary aneurysms, and central nervous system involvement. All patients have elevated levels of EBV DNA in their blood, which is well-correlated with clinical severity [19].
Morphologically, in patients with CAEBV-T/NK the lymph nodes exhibit paracortical hyperplasia with polymorphic and polyclonal lymphoid proliferation and large numbers of EBER positive cells. The liver exhibits portal or sinusoidal infiltration by small lymphocytes with no definite atypia (Fig. 2).
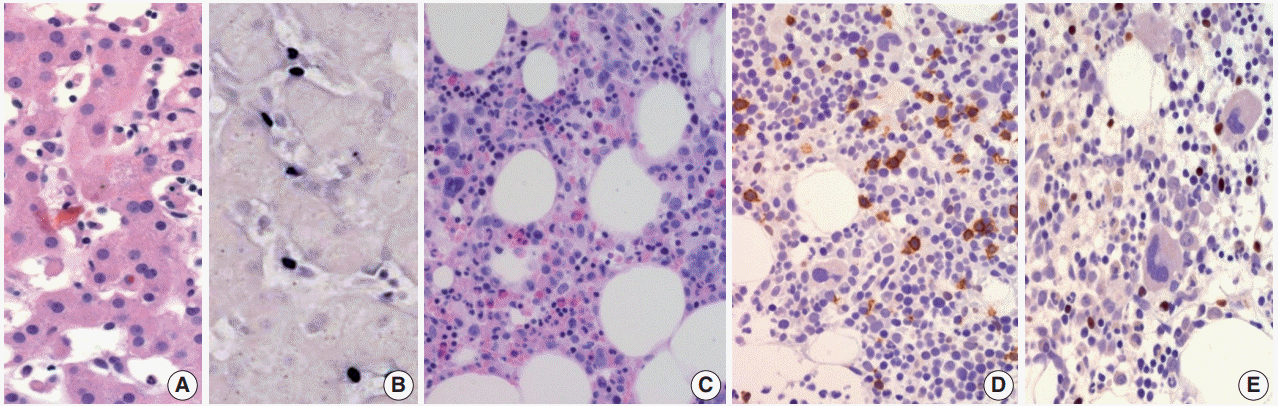
Chronic active Epstein-Barr virus (EBV) infection of a T-cell or natural killer cell type, systemic (Courtesy of Dr. Y.H. Ko). (A) A 21-yearold man presented with severe oral ulcer, recurrent pneumonia, thrombocytopenia, and elevated liver enzymes for 2 years. Liver biopsy reveals atypical T lymphocytes infiltrating the sinusoidal and hepatic lobules. (B) EBV-encoded small RNA (EBER) in-situ hybridization exhibits positive signals in these T cells. (C) Bone marrow biopsy shows small lymphocytic infiltrate. (D) CD3 is expressed in most lymphocytes. (E) EBER in-situ hybridization also shows positive signals in T cells.
Ohshima et al. [18] proposed a three-tier classification as follows: category A1 is polymorphic LPD with polyclonal proliferation of EBV-infected T cells or NK cells; category A2 is polymorphic LPD with monoclonal T/NK cells; and category A3 is monomorphic LPD of monoclonal T cells.
Severe mosquito bite allergy
A severe mosquito bite allergy is a cutaneous manifestation of chronic EBV infection characterized by intense local skin symptoms, such as erythema, bullae, ulcers, and scarring. The systemic symptoms such as fever, lymphadenopathy, and liver dysfunction are developed after mosquito bites, vaccination, or injection.
The epidermis at the mosquito bite site exhibits necrosis and ulceration. The dermis reveals edema and infiltration of polymorphonuclear leukocytes, nuclear debris, and extravasated erythrocytes with fibrinoid necrosis of small vessels. The infiltrating small lymphocytes extend from the dermis to the subcutis in an angiocentric pattern. EBV-positive cells represent 3%–10% of infiltrating lymphocytes [20].
Hydroa vacciniforme-like LPD
Hydroa vacciniforme (HV)-like LPD is one of the cutaneous forms of CAEBV. It is initially described as an EBV positive polyclonal or monoclonal T/NK LPD, characterized by blistering photodermatoses in childhood and healed with vacciniform scarring [21]. It is clinically divided into two types. The classic type is a self-limited disease with vesicles on sun-exposed areas in adolescence or young adulthood. Severe HV-type tends to exhibit more extensive skin lesions and systemic manifestations of fever, hepatomegaly, serologic abnormalities, and peripheral NK lymphocytosis. The severe type of HV often progresses to EBV-associated NK/T-cell malignancy [22,23].
Morphologic findings of HV are epidermal reticular degeneration to spongiotic vesiculation with perivascular and periappendiceal lymphocytic infiltration with no definite cytologic atypia. Severe HV and HV-like T-cell lymphomas mimic those of classic HV, but the dermal infiltrates are more extensive and deeper, composed of variably atypical lymphocytes.
The immunophenotype of the classic HV is CD4+ or CD8+ T-cells, but the severe form/lymphoma exhibits predominantly CD8+ CTLs. The majority are αβ T cells. A few cases involve αβ T cells and rarely NK cells.
Systemic EBV-positive T-cell lymphoma
Systemic EBV-positive T-cell lymphoma of childhood and young adulthood is a fulminant illness of EBV-infected T cells with clonal proliferation and cytotoxic phenotype. The clinical manifestation of this lesion is a rapid clinical progression with multiple organ failure, sepsis, and death. A hemophagocytic syndrome is nearly always associated. Systemic EBV-positive T-cell lymphoma arising in patients with a history of CAEBV-T/NK develops in a median time of 35 months [24].
Hyperplasia of histiocytes and marked hemophagocytic syndrome are also noted with increased small T-cells in the bone marrow, spleen, and liver. The paracortical zone of the lymph node is expanded with the depletion of B-cell areas. The degree of cytologic atypia in EBV-positive lymphocytes is variable.
Extranodal NK/T cell lymphoma, nasal type
Extranodal NK/T cell lymphoma is a prototype of EBV-associated T-cell LPD, which is characterized by frequent necrosis, angiocentric growth, cytotoxic phenotype and a strong association with EBV. Since the nasal cavity is the most commonly involved site, the nasopharynx and upper aerodigestive areas including the nasal cavity disclose progressively destructive and ulcerative lesions or obstructive symptoms due to mass effects [25].
Extranasal NK/T cell lymphoma
Extranasal NK/T cell lymphomas frequently involve the skin, gastrointestinal tract, testis, and soft tissue. Most patients present at a higher stage with multiple areas of involvement [26].
The morphology of the involved sites is frequently ulcerated and necrotic. The cytologic composition varies ranging from small, medium, and large. Angiocentric growth accompanies diffuse necrosis, and vascular damage.
Aggressive NK cell leukemia
Aggressive NK-cell leukemia is a neoplasm of NK cells, which primarily involves peripheral blood and bone marrow. In contrast to conventional leukemia, the tumor cells may not be abundant in the peripheral blood and bone marrow. The average age of patients exhibiting this disorder is 39 years. The typical presentations include fever, hepatosplenomegaly, lymphadenopathy, and is complicated by hemophagocytic syndrome.
In histologic sections, there are diffuse, destructive and permeative infiltrates of monomorphic cells with a round to moderate rim of pale or amphophilic cytoplasm. Interspersed apoptotic bodies and zonal cell death are common. Angioinvasive and angiodestructive growth is also frequently noted. The clinical course is fatal.
EBV-positive nodal NK/T cell lymphoma
EBV-positive nodal T/NK cell lymphoma may involve a limited number of extranodal organs except for the nasal cavity, but the main bulk of the tumor is located in the lymph nodes. This entity is very rare with fewer than 100 cases being reported in the literature [27].
The lymph nodes exhibit a diffuse infiltration of pleomorphic, variable sized cells. The cytomorphology of the tumor cells are more commonly centroblastoid, often anaplastic or plasmacytoid with some RS-like cells being noted. Some cases show extensive necrosis, many apoptotic bodies, and angiocentric growth patterns. The immunophenotype is as follows: CD3+, CD8+, TIA+, and granzyme B+ [28].
The disease course is very aggressive, with a median survival of only 4 months.
CONCLUSION
Many new entities and concepts for EBV-positive LPDs have been added to the 2016 WHO classification, based on growing knowledge in the field of genetics and molecular virology. With this increased understanding of LPD the clinical and pathologic entities, we can perform EBV-ISH indicative of (1) clinically bordered between infection and neoplastic conditions; (2) past history of recurrent inappropriate immune response, especially in children/young adults or old age; and (3) a pathologically polymorphous inflammatory background (but not Hodgkin lymphoma). It helps that the final destination is the achievement of appropriate diagnosis and management of LPDs. Some entities are provisional and must wait for confirmation of additional data.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.