Progressive Familial Intrahepatic Cholestasis in Korea: A Clinicopathological Study of Five Patients
Article information

Abstract
Background
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of autosomal recessive liver diseases that present as neonatal cholestasis. Little is known of this disease in Korea.
Methods
The records of five patients histologically diagnosed with PFIC, one with PFIC1 and four with PFIC2, by liver biopsy or transplant were reviewed, and ATP8B1 and ABCB11 mutation status was analyzed by direct DNA sequencing. Clinicopathological characteristics were correlated with genetic mutations.
Results
The first symptom in all patients was jaundice. Histologically, lobular cholestasis with bile plugs was the main finding in all patients, whereas diffuse or periportal cholestasis was identified only in patients with PFIC2. Giant cells and ballooning of hepatocytes were observed in three and three patients with PFIC2, respectively, but not in the patient with PFIC1. Immunostaining showed total loss of bile salt export pump in two patients with PFIC2 and focal loss in two. Lobular and portal based fibrosis were more advanced in PFIC2 than in PFIC1. ATP8B1 and ABCB11 mutations were identified in one PFIC1 and two PFIC2 patients, respectively. One PFIC1 and three PFIC2 patients underwent liver transplantation (LT). At age 7 months, one PFIC2 patient was diagnosed with concurrent hepatocellular carcinoma and infantile hemangioma in an explanted liver. The patient with PFIC1 developed steatohepatitis after LT. One patient showed recurrence of PFIC2 after 10 years and underwent LT.
Conclusions
PFIC is not rare in patients with neonatal cholestasis of unknown origin. Proper clinicopathologic correlation and genetic testing can enable early detection and management.
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of autosomally recessive inherited diseases, manifesting as neonatal cholestasis of hepatocellular origin. PFIC presents as jaundice, pruritus, and failure to thrive in neonates and infants, and usually advances to liver failure [1,2]. PFIC was first described in an Amish kindred and called Byler disease [3]. Molecular analysis showed distinct genetic mutations, classifying PFIC into three types, with all three characterized by a deficiency of bile transporters in hepatocytes. PFIC1 is characterized by mutations in the ATP8B1 gene, resulting in a deficiency in an aminophospholipid flippase that plays a role in maintaining plasma membrane integrity; PFIC2 is characterized by mutations in the ABCB11 gene (ATP-binding cassette family B, member 11), which encodes a bile salt export pump (BSEP); and PFIC3 is characterized by mutations in the MDR3 (class III multidrug resistance p-glycoprotein) gene, which encodes a flippase required for biliary phosphatidylcholine secretion [4-7].
PFIC1 and PFIC2 are both characterized by low serum γ- glutamyl transpeptidase (GGT) concentrations and direct hyperbilirubinemia, findings that are useful for their differential diagnosis from other neonatal cholestastic liver diseases [2]. Despite their distinct clinicopathologic features, pathologists and pediatricians are unfamiliar with these diseases because they are rare, with an estimated incidence rate of 1:50,000 to 1:100,000 births [2].
In Korea, PFIC has not been analyzed in detail and its clinicopathological course after diagnosis has not yet been reported. The present study therefore investigated the pathologic features, mutation profiles, and clinical courses of a series of patients at a single center diagnosed with PFIC.
MATERIALS AND METHODS
Ethical approval
This study was approved by the Institutional Review Board of Asan Medical Center, which waived the requirement for informed consent (IRB No. 2017-0703).
Patients and samples
From 2008 to 2014, five patients were diagnosed with PFIC at Asan Medical Center, Seoul, Republic of Korea. Diagnosis was based on clinical findings, histological and ultrastructural features of liver biopsy or liver transplantation (LT) samples, and mutation profiles. Clinical information, including age at onset, sex, chief complaints, and laboratory data, were retrieved from patients’ electronic medical records. Informed consent for liver biopsy and DNA analysis in all patients was obtained from the patients’ parents. No patients were lost to follow-up and the median follow-up time was 53 months (range, 36 to 102 months).
Histopathological analysis
Fifteen specimens from four liver biopsies, eight posttransplant liver biopies, and three explanted livers were histopathologically analyzed. All specimens were fixed in 10% neutral formalin and processed for hematoxylin-eosin and Masson trichrome staining to analyze histology and the stage of fibrosis. Specimens for electron microscropy were fixed in 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) for 48 hours, and postfixed in 1% osmium tetroxide. Ultrathin sections were cut, stained with uranyl acetate, and examined using a transmission electron microscope.
Samples were immunohistochemically stained with antibodies to cytokeratin 19 (1:200, DAKO, Glostrup, Denmark) and BSEP (1:1,000, Sigma Aldrich, St. Louis, MO, USA) to assess bile duct damage and BSEP protein expression, respectively, using a Benchmark XT auto immunostainer (Ventana Medical System, Tucson, AZ, USA) with OptiView DAB IHC Detection kit (Ventana Medical System) according to the manufacturer’s instructions. Histopathological features were carefully reviewed by three pathologists (E.Y., S.A.H., and H.J.K.) specializing in liver diseases, and the absence or presence of ballooning degeneration, giant cell transformation, and ductular reaction, as well as the extent of bile plugs, was determined. Lobular and portal based fibrosis were also analyzed, according to the Korean Society of Pathologists Scoring System [8,9]. Lobular fibrosis was graded as negative (no perivenular fibrosis), mild (perivenular fibrosis), moderate (perivenular and sinusoidal fibrosis), or severe (diffuse perivenular and sinusoidal fibrosis). Portal fibrosis was graded as no portal fibrosis, periportal fibrosis, septal fibrosis and cirrhosis.
Genetic test
Genomic DNA was isolated from peripheral blood leukocytes using Gentra Puregene blood kits (Qiagen, Hilden, Germany). Twenty-seven exons and exon-intron boundaries of ATP8B1 and 28 exons and exon-intron boundaries of ABCB11 were amplified by polymerase chain reaction (PCR) using GoTaq Polymerase (Promega, Madison, WI, USA), and the products were directly sequenced using BigDye Terminator kits v.3.1 (Applied Biosystems/Life Technologies, Waltham, MA, USA). The PCR products were electrophoresed using an ABI3130x1 Genetic Analyzer (Applied Biosystems/Life Technologies), and DNA sequences were compared with GenBank (http://www.ncbi.nlm.nih.gov) reference DNA sequences: NT_0025028.14 and NT_005403.17 for genomic sequences of ATP8B1 and ABCB11, respectively; and NM_005603.4 and NM_003742.2 for ATP8B1 and ABCB11 mRNAs, respectively. In silico prediction of the effects of missense variants in ATP8B1 and ABCB11 were performed using Poly-Phen-2 (http://genetics.bwh.harvard.edu/pph2) and Sorting Intolerance from Tolerant (SIFT; http://sift.jcvi.org).
RESULTS
Clinical and laboratory findings
The clinical characteristics of the five patients with PFIC are listed in Table 1. One was diagnosed with PFIC1 and four with PFIC2. Four were female and one was male. Age at PFIC onset was birth in one patient with PFIC1 and three with PFIC2, and 45 days in a fourth patient with PFIC2. Four patients were Korean and one was Arabian. The Arabian girl had been previously diagnosed with PFIC2 and underwent LT at another hospital. Jaundice was common to all five patients (100%). Only one PFIC2 patient had pruritus in addition to cholestasis. The median total bilirubin and direct bilirubin concentrations at the time of diagnosis were 19.6 mg/dL (range, 10.0 to 24.4 mg/dL; normal range, 0.2 to 1.2 mg/dL) and 15.0 mg/dL (range, 5.6 to 21.0; normal range, < 0.5 mg/dL), respectively. All patients had low or normal GGT (median, 30.0 mg/dL; range, 15.0 to 57.0 mg/dL; normal range, 8 to 61 mg/dL). Four patients, one with PFIC1 and three with PFIC2, underwent LT, followed by allograft liver biopsies performed two or three times.

Clinical and laboratory findings in five patients with PFIC
Histopathological findings
Histopathological features of the five patients with PFIC are shown in Table 2. All had intrahepatic cholestasis, but the patterns of cholestasis were different in patients with PFIC1 and PFIC2. The main finding in the patient with PFIC1 was bland lobular cholestasis with bile plugs (Fig. 1A), whereas the main findings in the patients with PFIC2 were canalicular, periportal, and/or cholangiolar cholestasis (Fig. 1B). Ballooning degeneration of hepatocytes (Fig. 1C) and giant cell transformation (Fig. 1D) were observed in three and three patients with PFIC2, respectively. Small cell changes in hepatocytes were observed only in the PFIC1 patient (Fig. 1A). Lobular activity ranged from none to severe, whereas portal activity was mild in the four patients with PFIC2 (75%). Ductular reactions were observed in the one patient with PFIC1 and in four with PFIC2. The stage of fibrosis varied in patients with PFIC2, with one having cirrhosis, two having periportal fibrosis, and one having no portal fibrosis. The PFIC1 patient had periportal fibrosis. Severe lobular fibrosis was observed in three PFIC2 patients (Fig. 2A), and mild fibrosis in one PFIC1 and one PFIC2 patient (Fig. 2B). Electron microscopy showed coarse and granular bile, called Byler’s bile, in the one PFIC1 patient (Fig. 3A), and amorphous bile in four patients with PFIC2 (Fig. 3B).

Pathologic features of five patients with PFIC
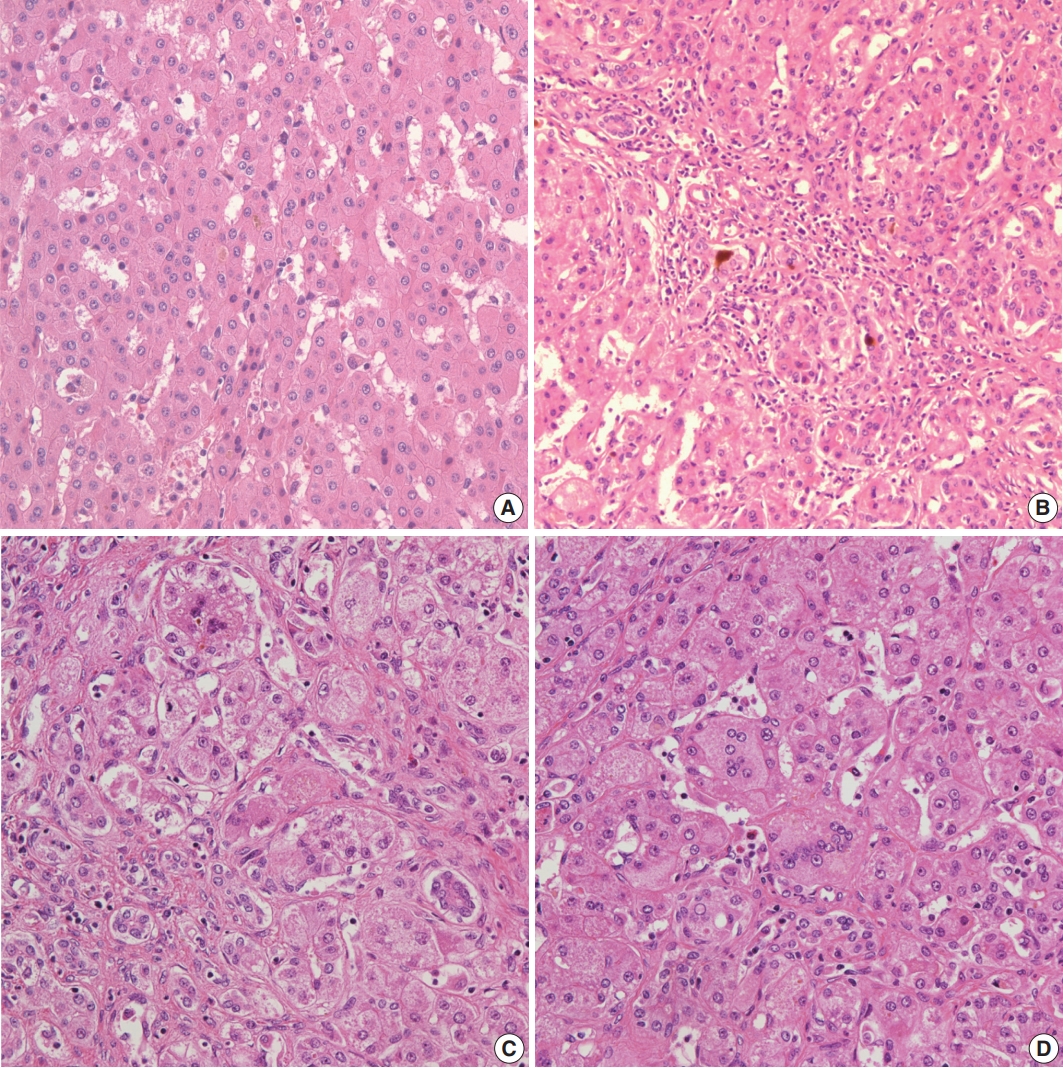
Histologic findings in patients with progressive familial intrahepatic cholestasis (PFIC). (A) Bland canalicular cholestasis and small cell change of hepatocytes with lobular disarray in our PFIC-1 patient. Bile duct proliferation with cholangiolar cholestasis (B), ballooning change (C), and giant cell transformation of hepatocytes (D) in patients with PFIC-2.
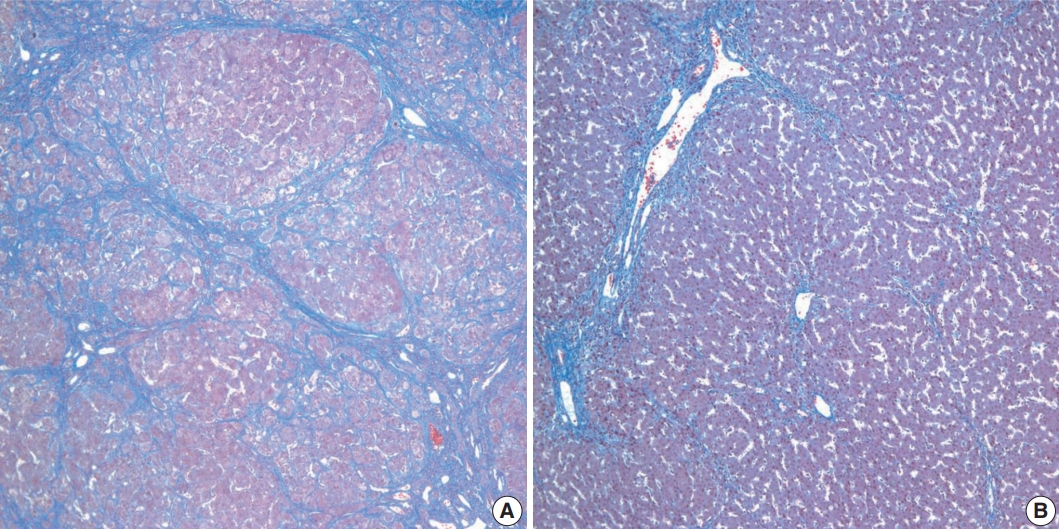
Stages of fibrosis in patients with progressive familial intrahepatic cholestasis (PFIC). (A) Cirrhosis with diffuse lobular fibrosis in a PFIC2 patient, as shown by Masson trichrome staining. (B) Periportal fibrosis with mild lobular fibrosis in the one patient with PFIC1.
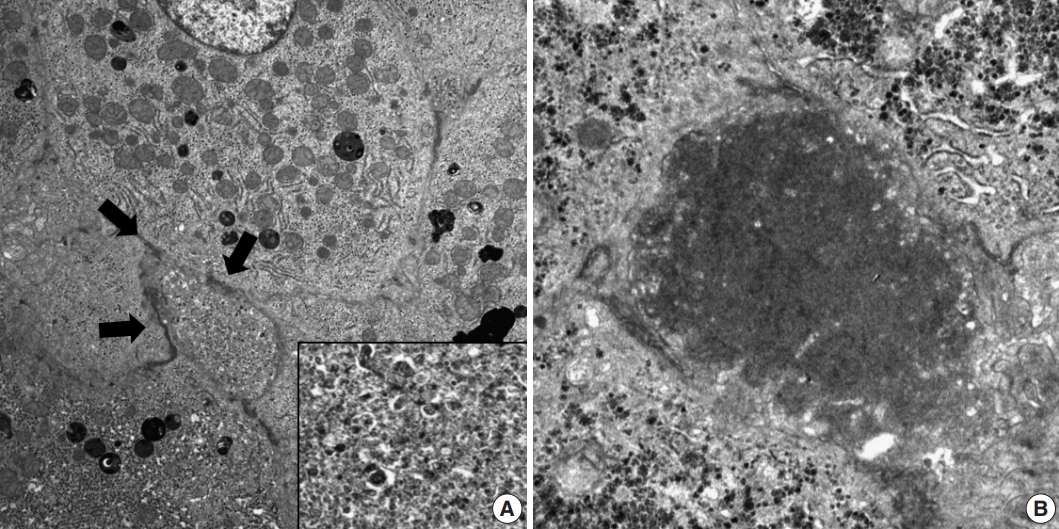
Electron microscopic findings in patients with progressive familial intrahepatic cholestasis (PFIC). (A) Dilated canaliculi (arrows) with coarse granular bile in the PFIC1 patient. (B) Amorphous and dense bile in PFIC2 patients.
BSEP immunostaining
BSEP immunohistochemical staining in patient with one PFIC1 showed normal canalicular expression (Fig. 4A). PFIC2 showed focal loss of BSEP expression in two patients (Fig. 4B) and total loss of BSEP expression in two patients (Fig. 4C), with the latter having an ABCB11 gene mutation.
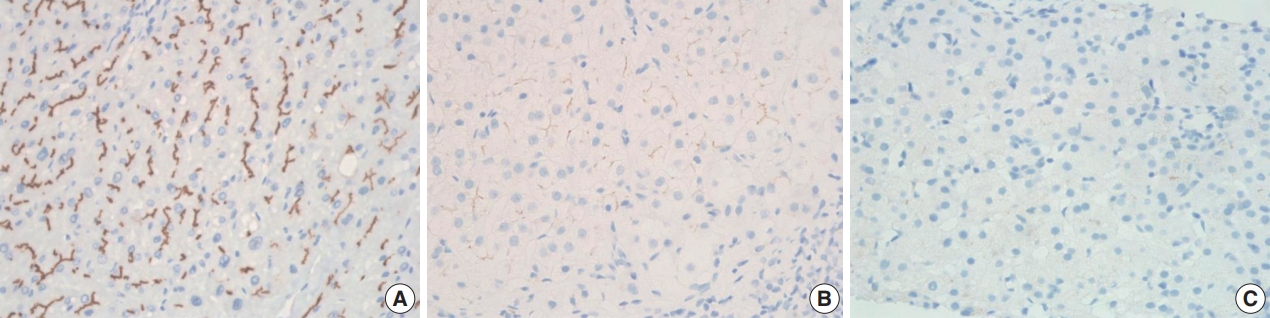
Immunohistochemical staining for bile salt export pump: normal canalicular expression (A), focal loss (B), and total loss (C).
Genetic mutation analysis
Genetic testing (Table 3) showed a nonsense mutation, c.1753G > T (p.E585*), in ATP8B1 in the one patient with PFIC1. Genetic data were available from all PFIC2 patients. Two patients had mutations in ABCB11, with one having a missense mutation, c.1907A > G (p.E636G), and the other having a nonsense mutation, c.1416T > A (p.Y472*). No mutations on ABCB11 gene were identified in two cases of PFIC2. In addition, the PFIC1 patient had a missense mutation, c.2246T > C (p.L749P) (c.2246T > C) in ATP8B1, and one PFIC2 patient had a missense mutation, c.2594C > T (p.A865V) (c.2594C > T) in ABCB11, with Poly-Phen-2 (http://genetics.bwh.harvard.edu/pph2) and SIFT (http://sift.jcvi.org) predicting that these missense mutations would lead to functional derangements of their respective proteins. These novel mutations could not be validated to exclude polymorphisms.

Results of genetic testing and BSEP immunostaining
Clinicopathological course
An abdominal computed tomography scan in one PFIC2 patient before LT showed multiple enhancing nodules and masses in both lobes of the liver. These were histologically found to be hepatocellular carcinomas and multiple infantile hemangiomas. Recurrence following LT was identified in the one Arabian PFIC2 patient. This patient experienced malnutrition and intractable jaundice 12 years after LT. Laboratory tests showed that, despite markedly elevated asparate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, and direct bilirubin concentrations, her GGT stayed in the normal range. Liver biopsy showed canalicular and periportal bile plugs, as well as diffuse giant cell and ballooning changes in hepatic lobules. Masson trichrome staining showed periportal fibrosis, and BSEP immunostaining showed complete loss of BSEP protein from the canalicular membrane.
The one patient diagnosed with PFIC1 did not show the expected recovery of AST and ALT concentrations after LT. A posttransplant liver biopsy showed severe fatty changes, ballooning degeneration, and mild lobular activity accompanied by sinusoidal and perivenular fibrosis. The final diagnosis was steatohepatitis, a frequent complication of LT in PFIC1 patients. One PFIC2 patient each developed acute cellular rejection and ischemic liver damage. All patients were alive at the last follow-up.
DISCUSSION
Although PFIC is a relatively well-documented disease worldwide, there have been only two case reports on PFIC in Korean patients. Identification of a mutation in an associated gene is not mandatory for diagnosis of PFIC, but is an important diagnostic tool [1]. However, clinical segmentation remains useful because, although PFIC has been reclassified according to molecular criteria, genetic testing is not always readily available. To our knowledge, this is the first study describing overall features of patients in Korea with PFIC.
PFIC is a heterogeneous group of liver diseases, but laboratory findings are similar in PFIC1 and PFIC2 patients, such as low GGT despite direct hyperbilirubinemia. These results suggest that hyperbilirubinemia in PFIC is associated with bile excretion by hepatocytes. By contrast, common forms of neonatal cholestasis are characterized by increased GGT, usually from damage to the bile ducts caused by the detergent effect of bile salts [10,11]. Although PFIC is unfamiliar to general pediatricians and pathologists, it is not a rare disease, accounting for 10%–15% of patients with neonatal cholestasis of unknown etiology. Knowledge of laboratory findings characteristic of PFIC narrows the differential diagnoses, enabling early diagnosis of PFIC in patients with intrahepatic cholestasis.
Some histopathologic features differ in PFIC1 and PFIC2 [12]. For example, PFIC1 is characterized by a bland lobular cholestasis with hepatocellular and canalicular patterns, as well as small cell changes in hepatocytes in early life biopsies [13]. Because our PFIC1 patient showing small cell change was 2 years old at the time of LT, the presence of small cell change is not limited to infancy but appears over time. Giant cell transformation of hepatocytes and ballooning change with cholestasis were major histologic features in PFIC2 patients, although the amount and distribution of giant cells and ballooning changes vary [14].
In contrast to PFIC2, abnormalities of the interlobular bile ducts are not observed in PFIC1 [2]. However, ductular reactions may appear in the latter, indicating disease progression [12]. The ductular reaction in our one PFIC1 patient may reflect her relatively older age.
Liver fibrosis is an early event of PFIC, with the grade and pattern of fibrosis influenced by patient age and type of PFIC. Lobular fibrosis may develop in PFIC1 and PFIC2, whereas portal fibrosis is a predominant feature of PFIC2 [15]. Portal fibrosis may be more associated with ductular reaction and portal pathology of PFIC2.
Electron microscopy has shown that bile properties differ in PFIC1 and PFIC2. Coarse granular bile, called Byler’s bile, is a distinct characteristic of PFIC1 and was used to diagnose our patient [16]. By contrast, PFIC2 is characterized by nonspecific fine bile located within dilated canaliculi, similar to findings in patients with intrahepatic cholestasis of various etiologies. Electron microscopic examination should therefore be included in the differential diagnosis of pediatric patients with cholestatic liver diseases.
About 80 and 100 genetic mutations have been identified in PFIC1 and PFIC2 patients, respectively [17,18]. Despite PFICs being autosomal recessive hereditary diseases, compound heterozygous or homozygous mutations may be associated with structural and functional defects. More severe forms of PFIC are likely associated with homozygous frame shift and nonsense mutations, as well as large genomic deletion. By contrast, a milder form of PFIC, benign recurrent intrahepatic cholestasis, is more likely associated with heterozygous missense mutations [19].
Most point mutations in PFIC1 and PFIC2 are missense, nonsense, and splicing mutations. Structurally abnormal proteins in PFIC1 are due to frame shift (26%), splice site (18%), and nonsense (13%) mutations, as well as large genomic deletion (3%). Functionally deficient proteins with normal structure in PFIC1 are caused by missense mutations (38%) and small inframe deletions (3%) [19]. The PFIC1 patient in the present study was found to have two heterozygous mutations; one nonsense, c.1753G > T (p.E585*), and one missense, c.2246T > C (p.L749P), mutation. Heterozygous mutations have been associated with good prognosis and low penetrance in PFIC [4,13]. However, our PFIC1 patient required an LT at age 26 months, suggesting that heterozygous mutations could not guarantee a benign clinical course in PFIC1 patients.
We failed to detect ABCB11 mutations in two PFIC2 patients in the present study. Fewer than 10% of PFIC patients have no or monoallelic mutations. Mutations in these patients may be present in regulatory domains, untranslated regions, and introns, which cannot be tested by present methods [20]. Despite the absence of mutations or the presence of a single heterozygous ABCB11 mutation, the diagnostic sensitivity of clinical and pathologic findings with negative BSEP immunostaining is approximately 90% [18]. Therefore, the absence of ABCB11 mutations cannot exclude PFIC2.
Ursodeoxycholic acid (UDCA) is considered an initial treatment for patients with PFICs [21]. UDCA can resolve symptoms in PFIC1 patients, but PFIC2 patients generally respond poorly, suggesting that UDCA has uncertain effects on the progression of liver disease [22]. In early stages of PFIC, partial biliary diversion that causes an unloading of bile acid may delay LT [23]. LT is the most efficient and last therapeutic option in patients presenting with liver failure [24,25]. The selection criteria for LT candidates do not differ from those in patients with other liver diseases. Major indications for LT include end stage liver disease, concurrent hepatocellular carcinoma, and intractable pruritus with no response to biliary diversion. Although LT results in remission in 75%–100% of patients, regardless of PFIC type, specific complications and relapse of disease should be carefully considered prior to LT [24-26]. Patients with PFIC1 may experience exacerbation of extrahepatic manifestations, including chronic diarrhea and liver steatosis [27,28]. For example, our one PFIC1 patient experienced rapid development of steatohepatitis not observed in the explant liver.
In summary, PFIC is not infrequently encountered in practice. Early detection and management should be based on proper clinicopathologic correlation and mutation analysis. Because it is more cost-effective, BSEP immunostaining should be performed prior to genetic testing to determine the type of PFIC. Mutation analysis of the ABCB11 gene should be performed in patients with portal tract abnormalities and reduction or loss of BSEP expression on immunostaining. In addition, electron microscopy should always be considered for the diagnosis and typing of PFIC.
Notes
Author Contributions
Conceptualization: EY, SAH, HJK.
Data curation: SAH, SHO, KMK, HWY.
Methodology: SHO, KMK, HWY.
Supervision: EY.
Visualization: HJK, SAH, GHK.
Writing—original draft: HJK, SAH.
Writing—review & editing: EY
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.