Renal intravascular large B cell lymphoma: the first case report in Korea and a review of the literature
Article information

Abstract
Herein, we describe the first case of renal intravascular large B cell lymphoma in Korea occurring in a 66-year-old female. She presented with mild fever and dyspnea. On physical and laboratory evaluations, hemophagocytic lymphohistiocytosis was suspected, but the bone marrow biopsy results were unremarkable. During the work-up, massive proteinuria developed, which led to a renal biopsy. The renal architecture was relatively well-preserved, but the glomeruli were hypercellular with the infiltration of atypical, large lymphoid cells with increased nucleus-cytoplasm ratio and clumped chromatin. Similar cells were also present in the peritubular capillaries. The tumor cells exhibited membranous staining for CD20 and CD79a. After the diagnosis of intravascular large B cell lymphoma, the patient received rituximab-based chemotherapy under close follow-up.
Intravascular large B cell lymphoma (IVLBL) is a rare, aggressive hematologic neoplasm characterized by the selective proliferation of neoplastic lymphoid cells within the lumina of small or intermediate sized vessels [1]. Most cases occur in adults, with a median age of 67 years [1]. Men and women are equally affected [2]. Clinically, IVLBL is often called ‘oncologist’s great imitator’, since it can affect various organs and manifest diverse symptoms according to the types of organs involved [3]. Central nervous system (CNS) and skin involvements are relatively frequent. However, renal involvement is rare [4]. In addition, fever, hepatosplenomegaly, pancytopenia, or even multiorgan failure may occur [5].
In this report, we described the first case of renal IVLBL in Korea occurring in a 66-year-old female. We also briefly reviewed 42 cases of renal IVLBL that were previously published.
CASE REPORT
A 66-year-old female was admitted to the Hematology Division of Internal Medicine with the presentation of a fever of 38°C, cough, non-bloody sputum, and dyspnea, which had started a month previously. Her past medical history was unremarkable. Her blood pressure was 126/67 mm Hg, pulse rate was 79 beats per minute, and respiratory rate was 18 breaths per minute. The physical examination revealed no specific findings. Neither lymphadenopathy nor skin rashes were noted. The analysis of the complete blood cell count revealed anemia (hemoglobin, 7.1 g/dL) and thrombocytopenia (platelet count, 95×103/μL). Ferritin was 1,003.0 ng/mL. Serum haptoglobin level was normal and the Coombs test was negative. The C-reactive protein level was 86.7 mg/L and the lactate dehydrogenase level was 1,779 U/L. Blood urea nitrogen and serum creatinine levels were 2.8 mmol/L and 0.75 mg/dL, respectively. Serum aspartate transaminase was 31 IU/L and alanine transaminase was 10 IU/L. Total bilirubin level was mildly elevated (1.5 mg/dL). Antinuclear antibodies, anti-neutrophil cytoplasmic antibodies, antiglomerular basement antibody, and hepatitis B and C viral markers were all negative. Sputum, blood, and urine cultures revealed all negative results. Chest computed tomography (CT) showed mild bronchiolitis in the right upper lobe field without definite evidence of pulmonary thromboembolism. Abdominal CT revealed mild hepatosplenomegaly. On the positron emission tomography–CT, diffuse 18F-Fluorodeoxyglucose (FDG) uptake on the bilateral lungs, mild FDG uptake on the left lateral segment of the liver, and increased FDG uptake along the axial and appendicular bones were noted (Fig. 1). Clinically, hemophagocytic lymphohistiocytosis was suggested. A subsequent bone marrow biopsy revealed normocellular marrow without abnormal cell infiltration. Liver biopsy showed mild lobular hepatitis with Kupffer cell hypertrophy and increased hemophagocytosis.
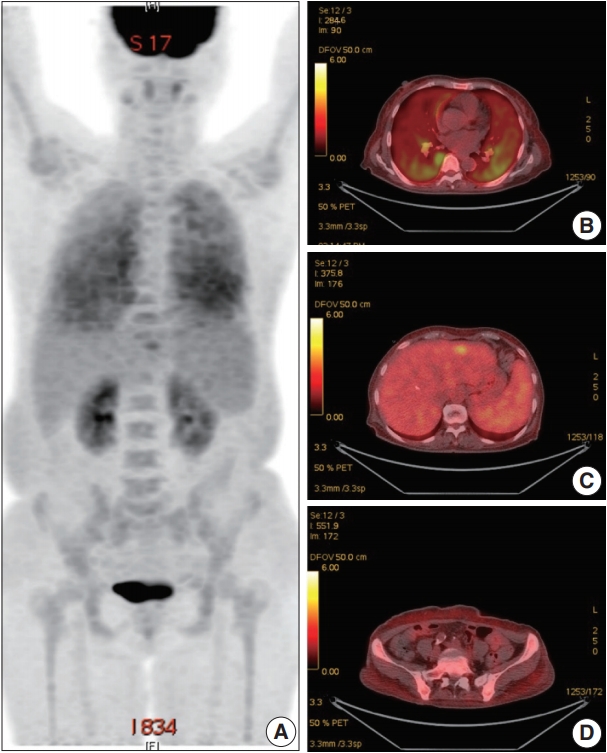
Positron emission tomography (PET) images (18F-Fluorodeoxyglucose PET). (A) Maximal intensity projection image. PET–computed tomography image of the lung (B), liver (C), and axial bones (D).
During further work ups, heavy proteinuria (4 g/day and urine protein-to-creatinine ratio of 4.94) suddenly developed. A renal biopsy was performed on the twenty-fourth hospital day. The biopsy core of the kidney contained nineteen glomeruli. Most of the glomeruli were mildly enlarged and hypercellular with infiltration of atypical large lymphoid cells in the capillary lumina. They exhibited a high nucleus-cytoplasm ratio with size variation, chromatin clumping, and inconspicuous nucleoli. Immunohistochemical stains demonstrated strong CD20 and CD79a membranous positivity with a Ki-67 proliferation index of 80% (Fig. 2). The tubulointerstitium was relatively wellpreserved, but the tumor cells were also present in some peritubular capillaries. On electron microscopy, the glomerular capillary lumen was obliterated by swollen endothelial cells and infiltrating atypical lymphoid cells (Fig. 3). The diagnosis of renal intravascular large B cell lymphoma was confirmed. The patient is currently receiving chemotherapy consisting of rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone (R-CHOP) with close follow-ups for three months.
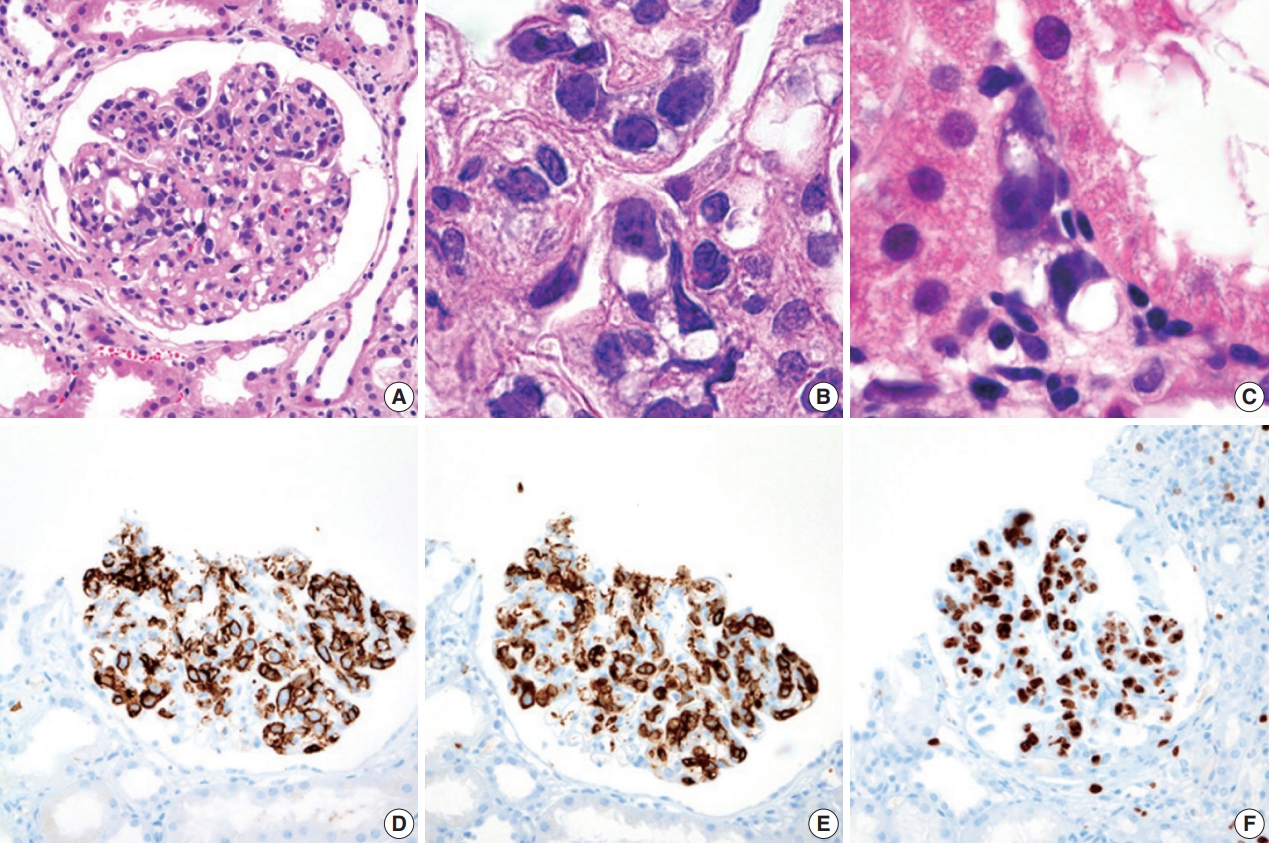
Histopathologic findings and immunohistochemical staining results of renal intravascular large B cell lymphoma. (A) Atypical large lymphoid cells are confined to the lumina of the glomerular capillaries. (B) On higher magnification, malignant lymphoid cells exhibited nuclear size variation, chromatin clumping, and scant cytoplasm. (C) Some atypical lymphoid cells are scattered in the peritubular capillaries. On immunohistochemical staining, these atypical lymphoid cells demonstrated strong membranous positivity for CD20 (D) and CD79a (E). (F) The Ki-67 proliferation index was 80%.
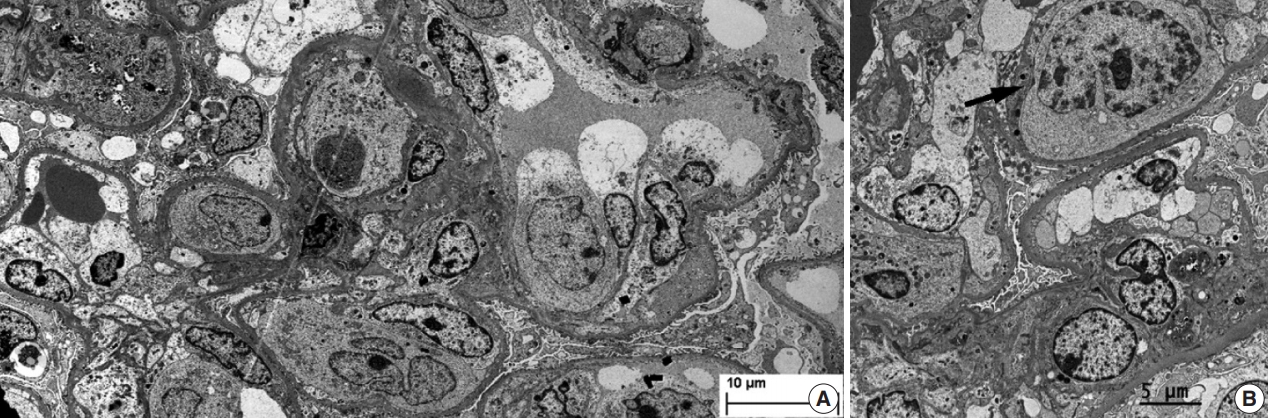
Representative electron microscopy of the kidney. (A) Atypical large lymphoid cells are present in the glomerular capillary lumen, and the podocytes exhibit diffuse foot process effacement (× 1,500). (B) On high power magnification, the atypical large lymphoid cell (arrow) shows prominent nucleoli and chromatin clumping (×3,000).
DISCUSSION
IVLBL is a rare type of aggressive, non-Hodgkin B cell lymphoma that is confined to small to medium sized vessels. Clinically, two major patterns have been recognized [6]. The first pattern, called the ‘Classic form’, is prevalent in Western populations and usually demonstrates skin and CNS involvement and related symptoms. The second pattern, so called the ‘Asian variant’, is characterized by hemophagocytic syndrome, fever, multiorgan failure, hepatosplenomegaly, and bone marrow involvement. Our case exhibited clinical features of the ‘Asian variant’ with fever, hepatosplenomegaly, and nephrotic range proteinuria.
Renal involvement of IVLBL is infrequent. Forty-three cases, including the present case, have been reported in English literature (Table 1). There were 19 males and 21 females, ranging from 35 to 85 years of age (mean age, 60.5 years). The clinical signs of renal involvement were proteinuria (90.0%), some with nephrotic range (21.0%), renal failure (60.5%), and enlargement of the kidney upon radiologic evaluation (34.3%). Fifty percent of the cases also exhibited extrarenal involvement. Bone marrow involvement was identified in three out of 16 cases. Hemophagocytosis was observed in four cases, including the present case. All four cases occurred in Asians.

Clinicopathologic characteristics of renal intravascular large B cell lymphoma
Histologically, malignant lymphoid cells demonstrate generally large, vesicular nuclei, one or more nucleoli, and scanty cytoplasm [1]. These cells usually express pan B cell markers including CD20, CD79a, and bcl-2, bcl-6, MUM-1, and variable expressions of CD5 and CD10 [7]. Although glomerular hypercellularity can be seen in hemophagocytic lymphohistiocytosis, cellular atypia and immunohistochemistry could differentiate malignant B lymphocytes from macrophages. Neoplastic cells lack surface molecules including CD29, CD54, and CD49d, facilitating the transvascular migration of the tumor cells. The aberrant expression of CXCR3 has also been described [8].
Of the 43 cases of intrarenal IVLBL with histologic descriptions, 35 cases exhibited the glomerular infiltration of tumor cells with or without peritubular and interstitial involvement. In four cases, the tumor cells were localized in peritubular capillaries without glomerular involvement. Our case showed glomerular and peritubular involvement. The microvascular infiltration and obliteration by tumor cells may be responsible for proteinuria in our case through podocyte and endothelial injuries [9].
IVLBL generally shows aggressive clinical courses, often worsened by the delay of diagnosis due to its nonspecific clinical presentation [10]. In terms of prognosis, at 6 months after diagnosis, 15 patients were alive and seven patients had died. Recently, rituximab-based chemotherapy has significantly improved clinical outcomes with a 3-year survival rate of 60%–80%. In addition, IVLBL only limited to the skin has been reported to exhibit better prognoses [11]. Our case is under rituximab-based chemotherapy for 3 months without further clinical deterioration.
In summary, we described a case of the renal involvement of IVLBL. Although the renal involvement of IVLBL is rare, this entity should be included as a differential diagnosis if unexplained hemophagocytic syndrome coupled with proteinuria persists.
Supplementary Materials
The Data Supplement is available with this article at https://doi.org/10.4132/jptm.2020.06.18.
Notes
Ethics Statement
This work was approved by a faculty research grant of Yonsei University College of Medicine (4-2020-0150). The patient provided written informed consent for the publication of associated data and accompanying images.
Author contributions
Conceptualization: HJJ. Data curation: MSK. Formal analysis: HJJ. Funding acquisition: HJJ. Investigation: MSK. Methodology: MSK, WIY, HRC. Project administration: HJJ. Resources: HJJ, WIY. Supervision: HJJ. Validation: HJJ, WIY, HRC. Visualization: HJJ, WIY, HRC. Writing—original draft: MSK. Writing—review & editing: HJJ, WIY, HRC. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.