Fibrocartilaginous mesenchymoma with an unusual location in the rib
Article information

Abstract
Fibrocartilaginous mesenchymoma is a rare bone tumor, with fewer than 35 cases reported in the literature since 1984. This tumor usually occurs in the long bones of children and adolescents. In the current case, the tumor affected a rib. A 17-year-old boy presented with a mass in the right fifth rib. Radiologic findings revealed an osteolytic mass with cortical destruction and calcification; en bloc resection was performed. The tumor showed three distinct histologic features: bland spindle cell proliferation, benign cartilage nodules, and epiphyseal plate-like enchondral ossification. The pathologic diagnosis was fibrocartilaginous mesenchymoma. The patient remains free of disease 1 year after the surgery. Pathological diagnosis of fibrocartilaginous mesenchymoma can be challenging, especially when the tumor occurs in an unusual site. When any fibro-osseous lesion with a cartilaginous component is encountered, the possibility of fibrocartilaginous mesenchymoma should be considered because of its locally aggressive behavior.
Fibrocartilaginous mesenchymoma (FM) is a rare, locally aggressive bone tumor, with fewer than 35 cases reported in the literature since 1984 when it was first reported by Dahlin et al. [1–3]. FM predominantly affects young patients with a median age of 13 years. This tumor principally occurs in long tubular bones and, infrequently, in the axial skeleton such as the iliac–pubic bones and vertebrae. Rib is an unusual location for FM, with only one published report [4]. Herein, we present a second case of FM arising in the rare anatomic site of the rib.
CASE REPORT
A 17-year-old boy was referred to the thoracic surgery department due to a mass located on the right side of the back. The patient had experienced intermittent pain and discomfort for several months at the time of admission, and localized swelling was noted on his back during physical examination. A chest radiograph showed a radiolucent lesion with cortical disruption in the posterolateral aspect of the right 5th rib. A computed tomography scan performed at the local clinic revealed an osteolytic mass with a juxtacortical extension (Fig. 1). Focal calcification was identified, but no periosteal reaction was observed. Overall radiologic findings suggested a benign tumor, including fibrous dysplasia (FD). En bloc resection was performed without a prior biopsy.
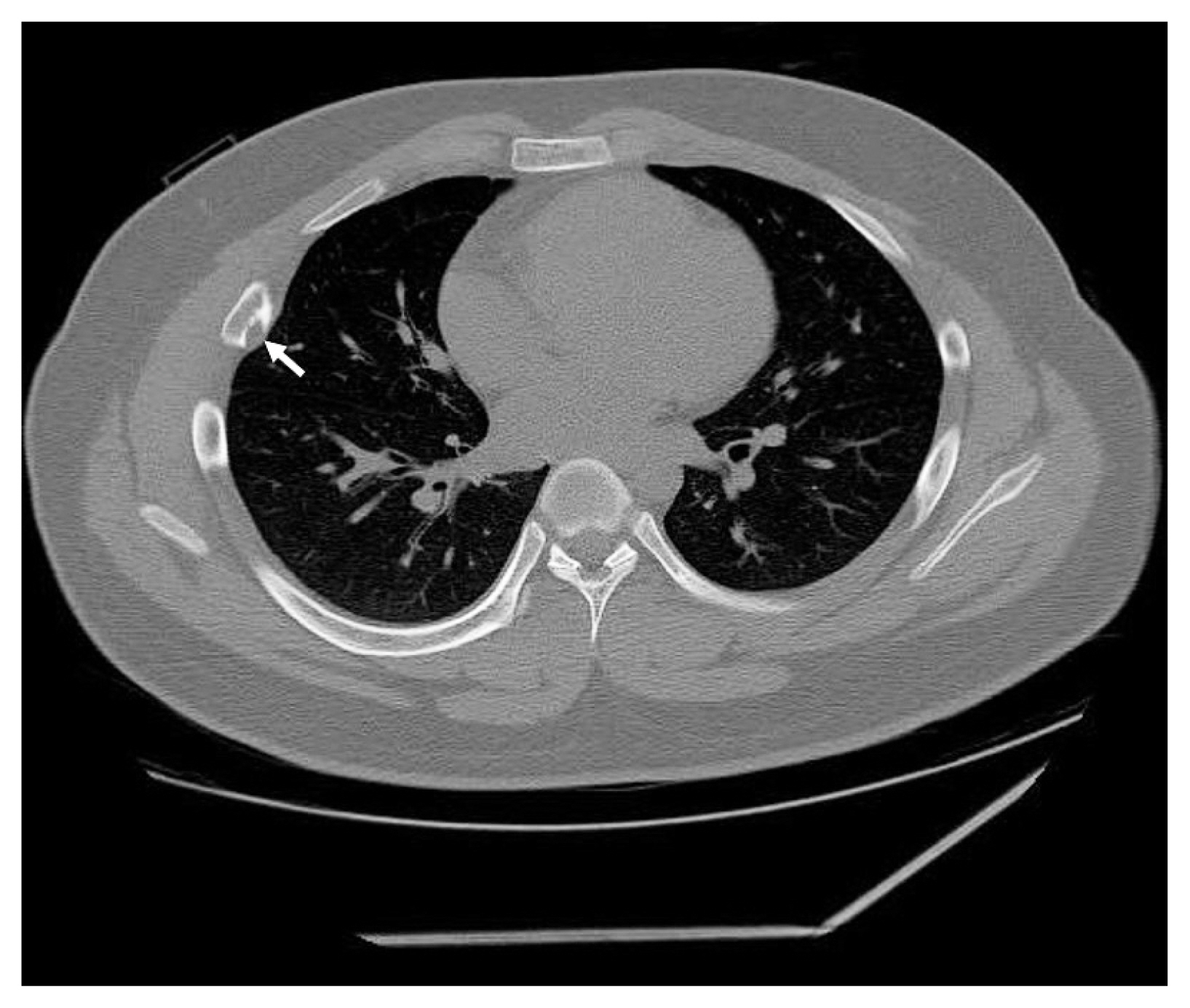
Computed tomography shows an osteolytic lesion (arrow) with calcification and juxtacortical extension at the posterolateral side of the right 5th rib.
The surgical specimen revealed a 3.1-cm-sized, ill-defined, eccentrically located, hard mass. The resected tumor was confined to the rib without expansion to the surrounding soft tissue, although the cortex had been partly destroyed. The cut surface of the tumor showed spotty areas of cartilaginous tissue (Fig. 2A). Histologically, the tumor was predominantly composed of hyaline cartilage nodules juxtaposed with hypocellular fibrous tissue (Fig. 2B). The cartilage islands throughout the tumor revealed low to intermediate cellularity but did not have any cytologic features of malignancy, such as nuclear atypia, multinucleation, mitosis, or myxoid changes (Fig. 2C). The hypocellular fibrotic area measured 1 cm in size, accounted for about 20% of the lesion, and consisted of benign spindle-to-stellate cells in a collagenous stroma without nuclear atypia or mitotic figures, suggesting low-grade sarcoma (Fig. 2D). The tumor was characterized by enchondral ossification at the periphery of the cartilage deposits, reminiscent of the epiphyseal growth plate or the cartilage cap of an osteochondroma (Fig. 2C). Newly formed bone was present and appeared as well-formed long and short trabeculae with prominent osteoblastic rimming. Multinucleated giant cells were not observed. Because the tumor was characterized as a fibro-osseous lesion with abundant cartilage nodules, the possibility of low-grade osteosarcoma, chondrosarcoma, and fibrocartilaginous dysplasia (FCD), a variant of FD, were considered. Immunohistochemical stains for murine double minute 2 (MDM2) and cyclin-dependent kinase 4 (CDK4) were negative in the spindle cells, excluding the possibility of low-grade osteosarcoma. Chondrosarcoma was unlikely because the cartilage islands were composed of benign chondrocytes without atypia, as described earlier. In addition, the lesion showed well-formed bone trabeculae and did not feature the characteristic, irregular, curvilinear trabeculae of woven bone without the osteoblastic rimming of FD or FCD. Epiphyseal growth plate-like enchondral ossification was another distinguishing feature, distinguishing it from FCD. A final diagnosis of FM was rendered based on the overall histologic findings. The patient was regularly followed every 3 months and is doing well 1 year after resection.
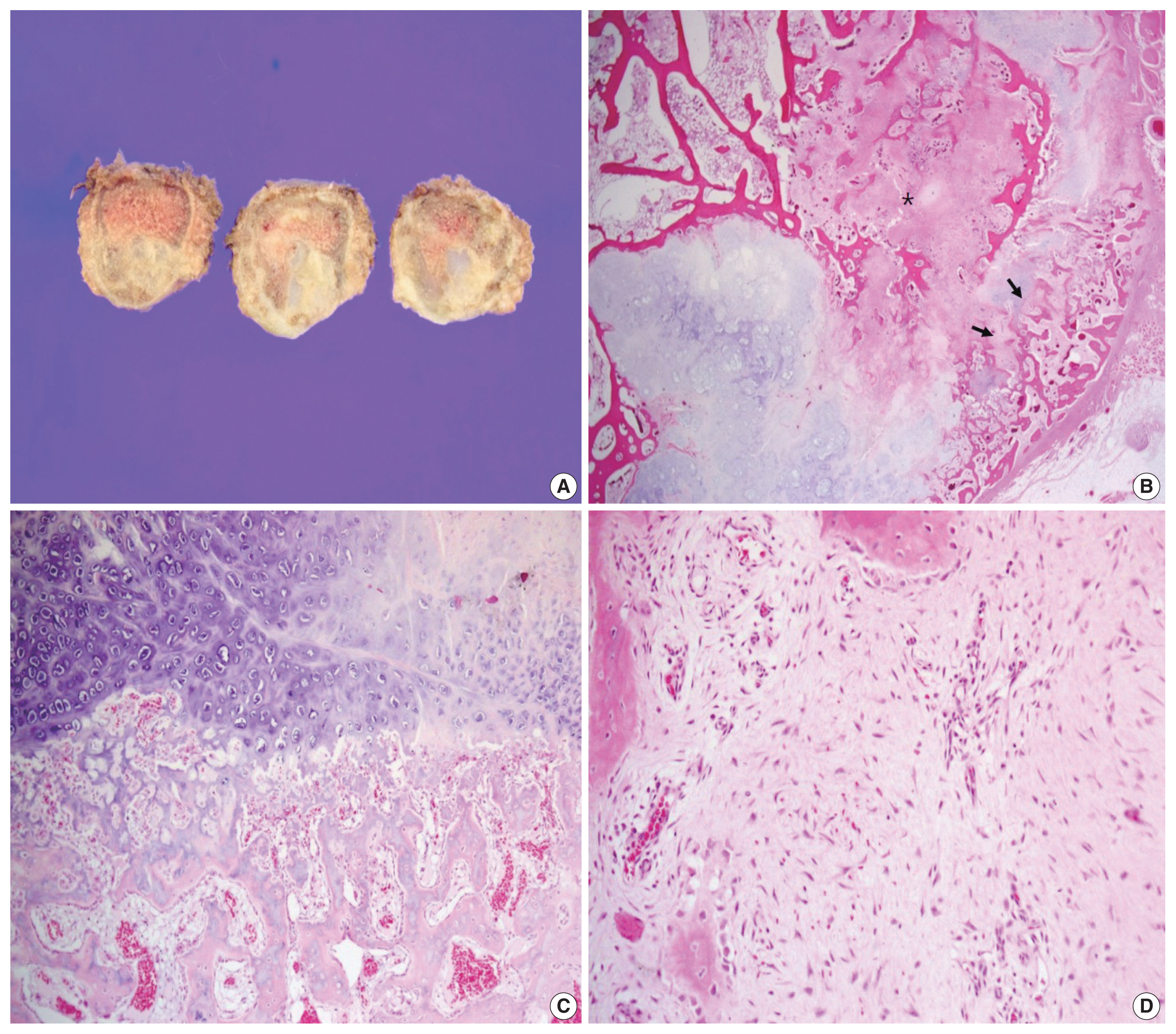
Histologic findings. (A) Gross examination reveals an ill-defined, eccentrically-located, hard mass with multifocal glistening bluish grey nodules corresponding to cartilage tissue. The cortex had been partially destroyed. (B) The tumor consists of multiple cartilage islands with a fibrocollagenous area (asterisk) and well-formed new bone trabeculae (arrows). (C) Epiphyseal plate-like enchondral ossification is observed at the periphery of the cytologically bland cartilage nodules. (D) A hypocellular fibrocollagenous lesion consisting of spindle and stellate cells without nuclear pleomorphism or mitosis is noted. Reactive woven bone formation with osteoblastic rimming is observed.
DISCUSSION
A rare neoplasm, FM was initially described as “a fibrocartilaginous mesenchymoma with low-grade malignancy” in 1984 by Dahlin et al. [1], who found unique lesions originally diagnosed as FCD but with different clinical, radiographic, and morphologic features. Since then, there have been debates in the literature regarding FM as a separate neoplasm or one that coincides with FCD [5–7]. However, several studies, particularly molecular analyses, have demonstrated that FM is not genetically related to FCD, which has a characteristic GNAS (guanine nucleotide-binding protein α-stimulating polypeptide) mutation [2]. Accordingly, the 5th edition of the World Health Organization classification of bone tumors newly listed FM as an independent tumor entity [8].
FM predominantly affects long bones, including the femur, tibia, and humerus; a few cases have been reported in rare locations, such as metatarsus, iliac–pubic bones, vertebrae, and ribs [2]. FM tends to affect young patients aged 3 months to 27 years (median age of 13 years), with a slight predominance of male patients. Radiologically, FM appears as an expansile osteolytic lesion with cartilaginous calcification and cortical destruction, and extension to soft tissue is not uncommon.
Histologically, FM is characterized by spindle cell proliferation in association with bland cartilage nodules and epiphyseal growth plate-like enchondral ossification. The morphology of spindle cells varies with respect to cellularity, nuclear pleomorphism, and mitotic figures, but it usually mimics low-grade spindle cell sarcoma. The occasional presence of multinucleated giant cells can be observed. Pathologic diagnosis can be challenging, particularly in small biopsies in which various fibro-osseous lesions with new bone formation must be considered.
The differential diagnoses include FCD, low-grade osteosarcoma, dedifferentiated chondrosarcoma, desmoplastic fibroma, and chondromesenchymal hamartoma of the chest wall when the tumor occurs in the ribs, as in the current case. FCD is a variant of FD and contains nodules of benign hyaline cartilage. Rare cases of FCD demonstrate enchondral ossification as in FM; however, FCD principally consists of bland spindle cells and characteristic, irregular, curvilinear trabeculae of woven bone, lacking osteoblastic rimming. Furthermore, GNAS mutations are detected in many cases of FCD, whereas no specific molecular aberrations have been found in FM [2]. Although the GNAS mutation was not tested in the present case, differentiation from FCD was not difficult due to the tumor’s overall histological characteristics. Finally, in contrast to FCD, FM is characterized clinically by a high propensity for local recurrence. The presence of epiphyseal plate-like cartilage is used to differentiate FM from desmoplastic fibroma and low-grade osteosarcoma. A molecular test for MDM2 amplification can be helpful to differentiate low-grade osteosarcoma from FM. Dedifferentiated chondrosarcoma is characterized by a low-grade cartilaginous tumor juxtaposed with a high-grade sarcoma. Cartilaginous nodules in dedifferentiated chondrosarcoma do not feature the epiphyseal growth plate-like appearance characteristic of FM. Moreover, dedifferentiated chondrosarcoma always shows high-grade sarcoma, whereas the spindle cells in FM commonly have a benign appearance. Chondromesenchymal hamartoma of the chest wall is histologically similar to FM in terms of a varying admixture of spindle cells, cartilage, and foci of epiphyseal growth plate-like enchondral ossification. However, chondromesenchymal hamartoma of the chest wall is a tumor of early infancy and occurs only in the rib; moreover, it has a prominent, aneurysmal, bone cystlike appearance that is not a feature of FM.
FM is a locally aggressive tumor with no distant metastasis reported thus far. Local recurrence occurs only in cases of incomplete removal, such as curettage or intralesional excision. Complete surgical excision with adequate margins is the treatment of choice for FM.
FM is a rare bone tumor that affects young patients and has a propensity for long tubular bones. We report a case of FM with an unusual location in the rib, which was successfully treated by en bloc resection. In cases of bone tumors that show characteristic histologic features of bland spindle cells, benign cartilage nodules, and epiphyseal plate-like enchondral ossification, physicians should consider the possibility of FM. Finally, FM is locally aggressive, so marginal resection or close follow-up for incomplete removal is required.
Notes
Ethics Statement
This study was approved by the institutional review board (IRB) of Kosin University Gospel Hospital with a waiver of informed consent (IRB No. 2019-07-037).
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.