SMARCA4/BRG1 protein-deficient thoracic tumors dictate re-examination of small biopsy reporting in non–small cell lung cancer
Article information

Abstract
Background
SMARCA4/BRG1 protein–deficient lung adenocarcinomas and thoracic sarcoma are recently described entities that lack distinctive histological features, transcription termination factor 1 (TTF1) reactivity, and actionable driver mutations. The current diagnostic path for small lung biopsies as recommended by the World Health Organization (WHO, 2015) is likely to categorize these as non–small cell carcinoma–not otherwise specified (NSCC-NOS). The present study attempts to define the subtle but distinctive clinicopathologic features of SMARCA4/BRG1 protein-deficient thoracic tumors; highlight their unique biology; and addresses the unmet need to segregate these using a new, tissue-proficient diagnostic pathway.
Methods
All lung biopsies and those from metastatic sites in patients with suspected advanced lung cancer and classified as NSCC-NOS as per WHO (2015) guidelines were subjected to BRG1 testing by immunohistochemistry. SMARCA4/BRG1 protein–deficient thoracic tumors were evaluated by an extended immunohistochemistry panel. Predictive biomarker and programmed death–ligand 1 testing was conducted in all cases.
Results
Of 110 cases, nine were found to be SMARCA4/BRG1 protein-deficient; six were identified as SMARCA4/BRG1 protein–deficient lung adenocarcinomas, and three were SMARCA4/BRG1 protein-deficient thoracic sarcomas. The histology ranged from poorly differentiated to undifferentiated to rhabdoid. None of the cases showed significant expression of TTF1 or p40, and no actionable mutation was identified.
Conclusions
It is difficult to separate BRG1-deficient lung adenocarcinomas and thoracic sarcomas based on morphology alone. We propose a diagnostic pathway for small biopsies of thoracic tumors to segregate these distinct entities so that they can be studied more efficaciously for new biomarkers and therapeutic options.
Switch/Sucrose non-fermenter (SWI/SNF) multiprotein complex is an ATP-dependent chromatin remodeling factor. One important and core constituent of this multiprotein complex is the brahma related gene 1 (BRG1) protein encoded by the SMARCA4 gene. The SMARCA4/BRG1 protein hydrolyzes ATP and provides energy for unspooling DNA from the histone octamer, allowing transcription to proceed [1–3]. The chromatin remodeling complex also plays an essential role in maintenance of stemness [4]. The pathogenesis and dedifferentiation of neoplasms in various organs are linked increasingly to chromatin remodeling by the SWI/SNF complex [5–7].
SMARCA4-inactivating mutations and consequent loss of functional SMARCA4/BRG1 protein are observed in many tumor types [6–8] and are observed in 8.43% of non–small cell lung cancers (NSCLC) [9–12]. Furthermore, SMARCA4/BRG1 protein–deficient thoracic sarcoma (SD-TS) is also recognized more frequently, primarily due to rising awareness of its existence [13–15]. There is uncertainty as to whether the histogenesis of SD-TS represents undifferentiated/dedifferentiated carcinomas or de novo genesis [16,17]. Overlapping histomorphology of SMARCA4/BRG1 protein–deficient lung adenocarcinoma (SD-LUAD), SD-TS, and other lung adenocarcinomas necessitates more exhaustive immunophenotyping than allowed with the current diagnostic pathway for small lung biopsy [18]. SMARCA4/BRG1 protein–deficient thoracic tumors (SD-TT) constitute a significant percentage of thoracic malignancies with reasonable fear of being inappropriately classified as non-small cell carcinoma–not otherwise specified (NSCC-NOS) and subjected to molecular testing. SD-TT are devoid of actionable molecular targets and need to be distinguished and studied separately to determine the best standard of care for these highly aggressive and rapidly lethal tumors and to save resources on unjustifiable predictive biomarker testing for NSCC-NOS. Moreover, SD-LUAD and SD-TS must be separated properly to retain histogenetic fidelity and to develop suitable therapeutic modalities.
Here, we describe a series of SD-TT and share a new diagnostic pathway to effectively segregate this distinct subset with proficient use of biopsied tissue and immunohistochemistry (IHC).
MATERIALS AND METHODS
Study place and study duration
The present study is an analysis of nine cases of SD-TT diagnosed at Rajiv Gandhi Cancer Institute and Research Center, New Delhi, from 1 September 2020 to 15 January 2021. Clinicoradiological details were obtained from the electronic medical records (EMR), and follow-up information was gathered either through EMR or via telephone.
Study population
The current World Health Organization (WHO) guidelines do not advocate extensive immunophenotyping beyond transcription termination factor 1 (TTF1) and p40 for classifying small biopsy specimens from suspected lung cancers and classify lung carcinomas as NSCC-NOS in the absence of both TTF1 and p40 [18]. SD-TT have been shown to be confined largely to TTF1-negative/low expression NSCC [10,11,19]. We reaffirmed these findings using two tissue microarrays of 25 samples of TTF1-negative and TTF1-positive NSCC subjected to BRG1 IHC testing. None of the TTF1-positive (moderate to strong nuclear expression) NSCCs showed loss of BRG1 expression, while eight of 25 cases of TTF1-negative NSCC showed loss of BRG1 expression. With similar findings noted in the literature, we applied additional IHC for BRG1 expression for this subset of lung cancers to identify SD-TT starting in September 2020. Nine such cases were identified to be BRG1 deficient. Of these, eight were newly diagnosed, and one (case number 4) underwent repeat biopsy for programmed death–ligand 1 (PD-L1) tumor proportion scoring after failure of multiple lines of cytotoxic therapy.
Immunohistochemistry
All lung biopsies and those from metastatic sites in patients with suspected advance lung cancers were classified per WHO (2015) guidelines [18] into NSCC-Adenocarcinoma (Ad Ca)/favor adenocarcinoma, NSCC–squamous cell carcinoma (SCC)/favor SCC, and NSCC-NOS using only p40 (clone BC28, ready-to-use [RTU], Zytomed Systems, Berlin, Germany) and TTF1 (clone-SP141, RTU, Ventana, Tucson, AZ, USA) IHC. Those classified as NSCC-NOS were tested for expression of SMARCA4/BRG1 protein (1:100, EPNCIR111A, Abcam, Cambridge, UK). Cases with loss of BRG1 protein were classified as SD-TT and further evaluated using an extended IHC panel of panCK (1:200, AE1, AE3, 5D3, Zytomed), cytokeratin 7 (CK7; 1:200, OV-TL 12/30, Thermo Fisher Scientific, Waltham, MA, USA), epithelial membrane antigen (EMA; 1:50, E29, Dako, Santa Clara, CA, USA), Hep Par 1 (OCHIE5, RTU, PathnSitu, Pleasanton, CA, USA), SALL4 (EP299, RTU, PathnSitu), SOX2 (1:100, SP76, Cell Marque, Darmstadt, Germany), CD34 (QBEnd10, RTU, Dako), synaptophysin (1:100, MRQ-40, Cell Marque), SMARCB1/INI1 (MRQ-27, RTU, Cell Marque), and E-cadherin (NCH-38, 1:100, Dako). Tissues were formalin-fixed, paraffin-embedded, sectioned to 4 μm thickness, and then fixed for 6–48 hours in neutral buffered formalin before conventional tissue processing and staining by IHC. Antigen retrieval at alkaline pH of 8.6 in Tris-EDTA buffer and further staining steps were performed on an automated IHC staining instrument (Benchmark XT, Ventana Medical Systems, Inc. [F. Hoffmann-La Roche Ltd.]). The chromogenic signal was generated using the Ventana UltraView universal 3,3′-diaminobenzidine (DAB) detection kit (Ventana Medical Systems, Inc. [F. Hoffmann-La Roche Ltd.]). All IHC stains were applied with appropriate positive controls. For IHC analysis, moderate to strong staining intensity in > 5% of tumor cells with nuclear (TTF-1, p40, SOX2, and SALL4), cytoplasmic (panCK, CK7, CD34, Hep Par 1, and synaptophysin), or membranous (EMA and E-cadherin) pattern was considered positive. BRG1 immunostaining that exhibited a null phenotype in tumor cells with strong nuclear staining in the stromal fibroblasts as well as the endothelial and inflammatory cells was deemed as lost and defined the SD-TT cohort.
Anaplastic lymphoma kinase 1 (ALK-1) protein expression was assessed using a U.S. Food and Drug Administration (FDA)–approved IHC assay employing anti-ALK (D5F3) rabbit monoclonal primary antibody with other proprietary components of the Ventana ALK assay on the Ventana Benchmark XT Autostainer (using the Ventana Optiview DAB and amplification kit).
PD-L1 expression testing was performed using rabbit Anti-Human PD-L1 monoclonal antibody (clone SP263) on the Ventana Benchmark XT, and partial or complete membranous expression of any intensity in ≥1% of tumor cells was considered significant.
DNA extraction and polymerase chain reaction
A predictive biomarker search was performed for epidermal growth factor receptor (EGFR) using the Qiagen EGFR Therascreen RGQ polymerase chain reaction (PCR) kit, an FDA-approved amplification refractory mutation system. Five sections of 4 μM each were collected in Eppendorf tubes by manual macro-dissection to enrich tumor fraction wherever necessary. DNA was extracted using the Qiagen DNeasy blood and tissue kit (Hilden, Germany). The DNA was quality checked on the Qubit fluorometer. Multiplexed reverse transcription PCR was carried out on a ROTORGENE thermal cycler (Qiagen) in eight tubes along with positive and no template controls. Interpretation was conducted per the manufacturer’s instructions.
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) analysis was performed on 4 μM formalin-fixed and paraffin-embedded tissue sections following a standardized protocol that included pretreatment (dewax/proteolysis), denaturation, probe application, and hybridization; application of 4′,6-diamidino-2-phenylindole (DAPI)/antifade solution; and analysis of slides using Leica fluorescent microscopy (DM6000B). ROS1 (ROS proto-oncogene 1, receptor tyrosine kinase) rearrangement was tested by FISH using a dual-color break-apart probe (ZytoLight Spec ROS1, ZytoVision, Germany) as previously described [20].
One case (case No. 1) was subjected to a targeted panel for multigene profiling using the Oncomine focus assay on the Ion Torrent Personal Genome Machine (Ion PGM, Thermo Fisher Scientific). Reaffirmation of the next-generation sequencing findings for copy number gain used FISH for c-MYC. The XL MYC BA spectral orange-labeled probe hybridizing proximal to the MYC gene region at 8q24.21 and a green-labeled probe hybridizing distal to the MYC gene region at 8q24.21 were applied (Metasystems Probes GmbH, Altlussheim, Germany). No centromeric probe was used. The number of c-MYC signals per cell was counted in 100 tumor cells and averaged. c-MYC copy number gain was defined as average copy number ≥ 3.0.
RESULTS
Clinical findings
A total of 110 cases of thoracic tumors was identified during this period, of which nine (8.1%) were found to be SMARCA4 deficient. Six cases (case Nos. 1–6) were SD-LUAD, and three (case Nos. 7–9) were SD-TS. Patient age ranged from 45–73 years, with a male to female ratio of 3.5:1. All cases of SD-LUAD had history of smoking, lung mass, bulky lymphadenopathy, and bony involvement. All cases of SD-TS had a significant history of smoking; two (case No. 7, case No. 9) had massive lung disease (Fig. 1A), while one had mediastinal disease (case No. 8) (Fig. 1B). All cases of SD-TS also had bulky lymphadenopathy. However, no bony involvement was noted in any SD-TS patients. The clinical features of SD-TT are summarized in Table 1.
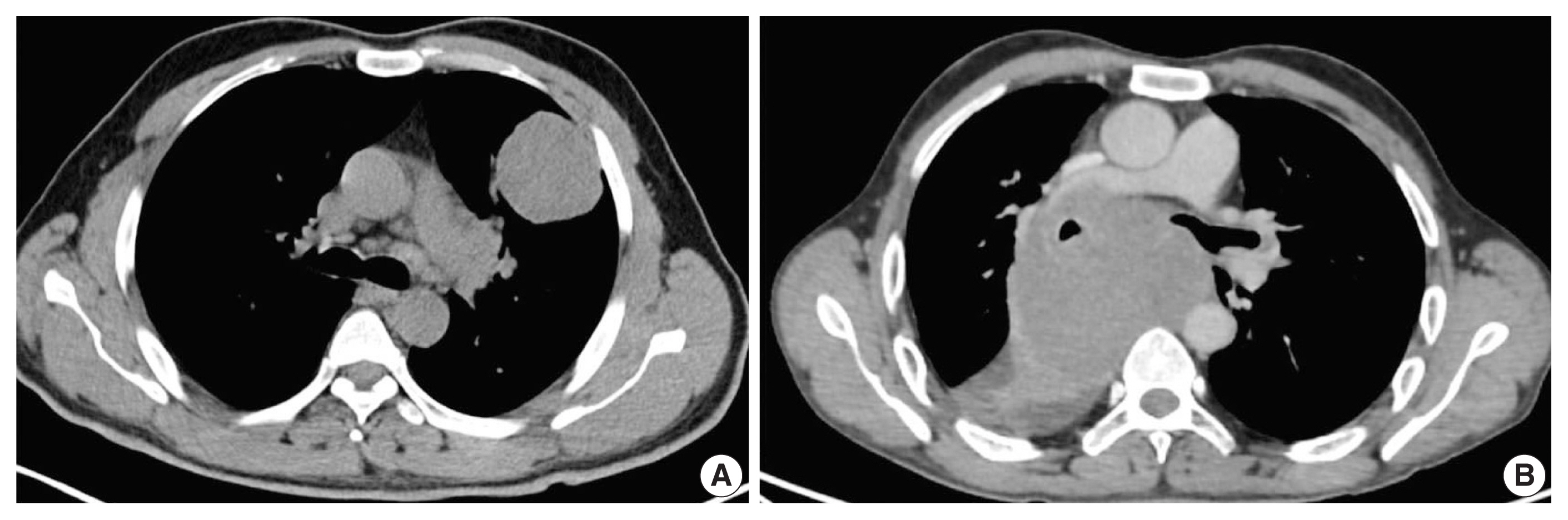
Axial computed tomography images of SMARCA4/BRG1 protein–deficient thoracic sarcoma (SD-TS). (A) Case No. 7 showed well defined intra-pulmonary mass on the left side, abutting the pleural margin. (B) Case No. 8 showed a well defined heterogeneously enhancing mediastinal-based mass splaying the carina and abutting the right pulmonary artery, encasing the right main bronchus without invasion.

Clinical features of SMARCA4/BRG1 protein-deficient thoracic tumors
Pathological findings in SD-LUAD
The histopathologic characteristics of SD-LUAD are summarized in Table 2 (case Nos. 1–6) and shown in Fig. 2A–D. All six cases of SD-LUAD had a solid pattern of growth in the biopsied material, while case No. 2 also showed a vague acinar pattern focally. One case (case No. 2) had an Indian file pattern of growth that resembled lobular carcinoma of breast (Fig. 2C); however, E-cadherin expression was intact, and none of the IHC markers for breast cancer (GATA3, estrogen receptor, and progesterone receptor) were positive. Case Nos. 4 and 6 showed multicell trabecular patterns (3–4 cell layer thick) reminiscent of hepatocellular carcinoma. All tumors were characterized by large constituent cells possessing eosinophilic to clear cytoplasm (Fig. 2A, B). Scattered populations of rhabdoid cells were seen in two cases (case Nos. 3 and 5) (Fig. 2D). Four cases (case Nos. 1, 2, 4, and 6) exhibited scattered cells with blue intracytoplasmic mucin, which were mucicarmine positive (Fig. 2E).

Histopathological profile of SMARCA4/BRG1 protein-deficient thoracic tumors
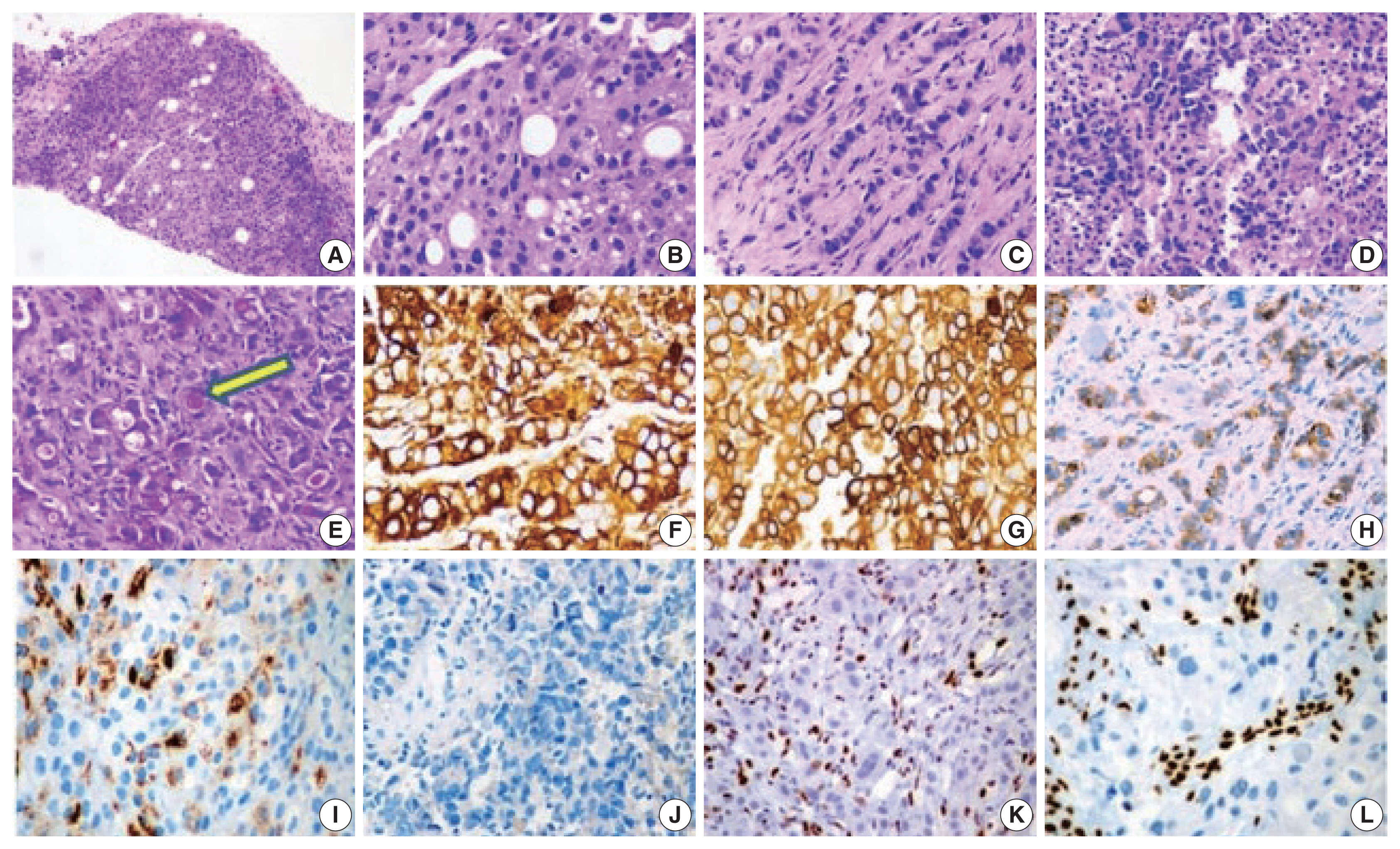
Histology and immunohistochemistry images of SMARCA4/BRG1 protein–deficient lung adenocarcinoma (SD-LUAD). (A) Scanner view of SD-LUAD. Note the solid pattern of growth with a sieved appearance. (B) The dry high-power view exhibits large cells with sharp margins and scattered large signet ring cells that are regularly observed in this tumor type. Also note the bubbly cytoplasm of many cells with indented nuclei. (C) The second case of SD-LUAD exhibited tumor cells arranged in an Indian file pattern. (D) The third case of SD-LUAD showed with highly inflamed background and a solid growth pattern comprised of large rhabdoid cells. (E) Tumor cells showed intra-cytoplasmic mucin (arrow) highlighted by mucicarmine. (F, G) Tumor cells showed strong cytokeratin (CK) and CK7 immunoreactivity, respectively. (H) SD-LUAD expressed BerEp4, supporting the adenocarcinomatous histogenesis. (I) All SD-LUAD expressed varying degrees of Hep Par 1. (J) Transcription termination factor 1 was universally absent. (K, L) No SD-LUAD exhibited BRG1 nuclear expression. Note the strong nuclear reactivity for BRG1 in the inflammatory cells.
The cytoplasmic margins of large polygonal cells were sharp and mimicked squamous morphology. The nuclei were large and irregularly contoured with cloudy chromatin. The rhabdoid population of tumor cells showed well-described, eccentric nuclei with prominent eosinophilic nucleoli and a cytoplasmic globule filled with hyaline content (Fig. 2D). Mitosis was intense. Apoptosis and wide swaths of necrosis were common. All our cases revealed inflamed stroma rich in lymphocytes and neutrophils (Fig. 2D). Neutrophilic emperipolesis was observed in three cases (Table 2).
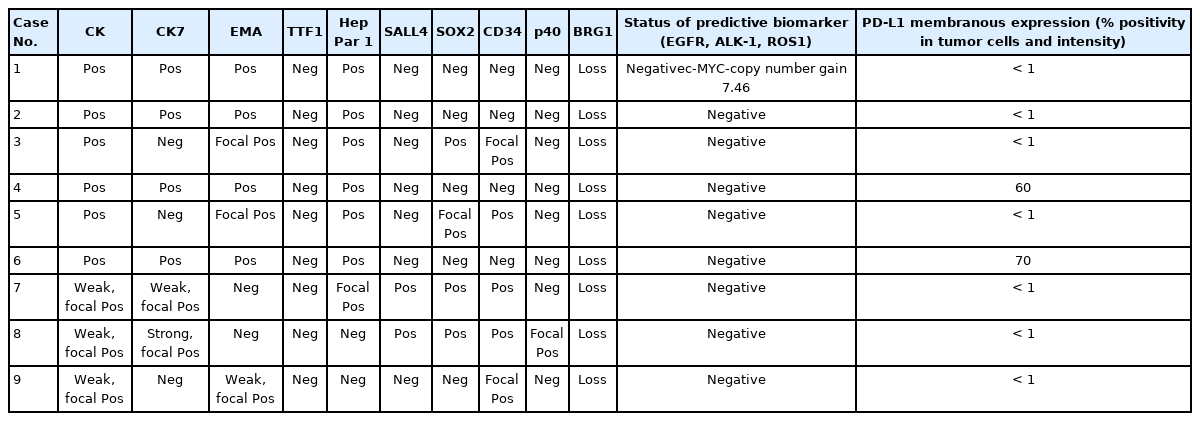
The IHC profile of SD-LUAD is summarized in Table 3 and shown in Fig. 2F–L. Cases Nos. 1, 2, 4, and 6 exhibited diffuse positivity for CK, CK7, and BerEp4 (Fig. 2F–H) and variably intense positivity for Hep Par 1 (Fig. 2I), but none of the cases was positive for SOX2, CD34, or SALL4. Diffuse positivity for CK7 and BerEp4 was consistent with adenocarcinoma appellation. Case Nos. 3 and 5 had strong CK and Hep Par 1 positivity along with focal positivity for two of three markers of stemness (SOX2, CD34, and SALL4), while both of them were negative for CK7 and BerEp4. Cases that showed focal expression of markers of stemness also had focal rhabdoid morphology.
Ancillary testing results of SMARCA4/BRG1 protein-deficient thoracic tumors
Pathological findings in SD-TS
The histopathologic characteristics of SD-TS are summarized in Table 2 (case Nos. 7–9) and shown in Fig. 3A–F. All three cases had a solid pattern of growth with pure rhabdoid morphology seen in two cases (case Nos. 7 and 9) (Fig. 3D–F). One case (case No. 8) had morphology similar to the features described in SD-LUAD (Fig. 3A–C). None of these cases showed areas of spindling, myxoid change, or any other feature that raised suspicion of sarcomatous histogenesis. Additional features noted in SD-LUAD such as inflamed stroma, neutrophilic emperipolesis, necrosis, brisk mitoses, and apoptosis were seen in these tumors (Table 2, Fig. 3D, E). On IHC, all cases had weak focal positivity for CK (Fig. 4A); two cases exhibited focal expression of CK7 (case Nos. 7 and 8) (Fig. 4B, C), and one case (case No. 9) showed weak focal expression for EMA. Hep Par 1 (Fig. 4D) and synaptophysin (Fig. 4H) were focally expressed in case No. 7, and p40 was focally expressed in case No. 8. Case Nos. 7 and 8 showed diffuse positivity for stem cell markers SALL4, SOX2, and CD34 (Fig. 4E–G) along with BRG1 loss (Fig. 4I), while case No. 9 had focal CD34 positivity in combination with BRG1 loss. SMARCB1/INI1 was intact in all cases. Table 3 and Fig. 4 show the IHC of SD-TS.
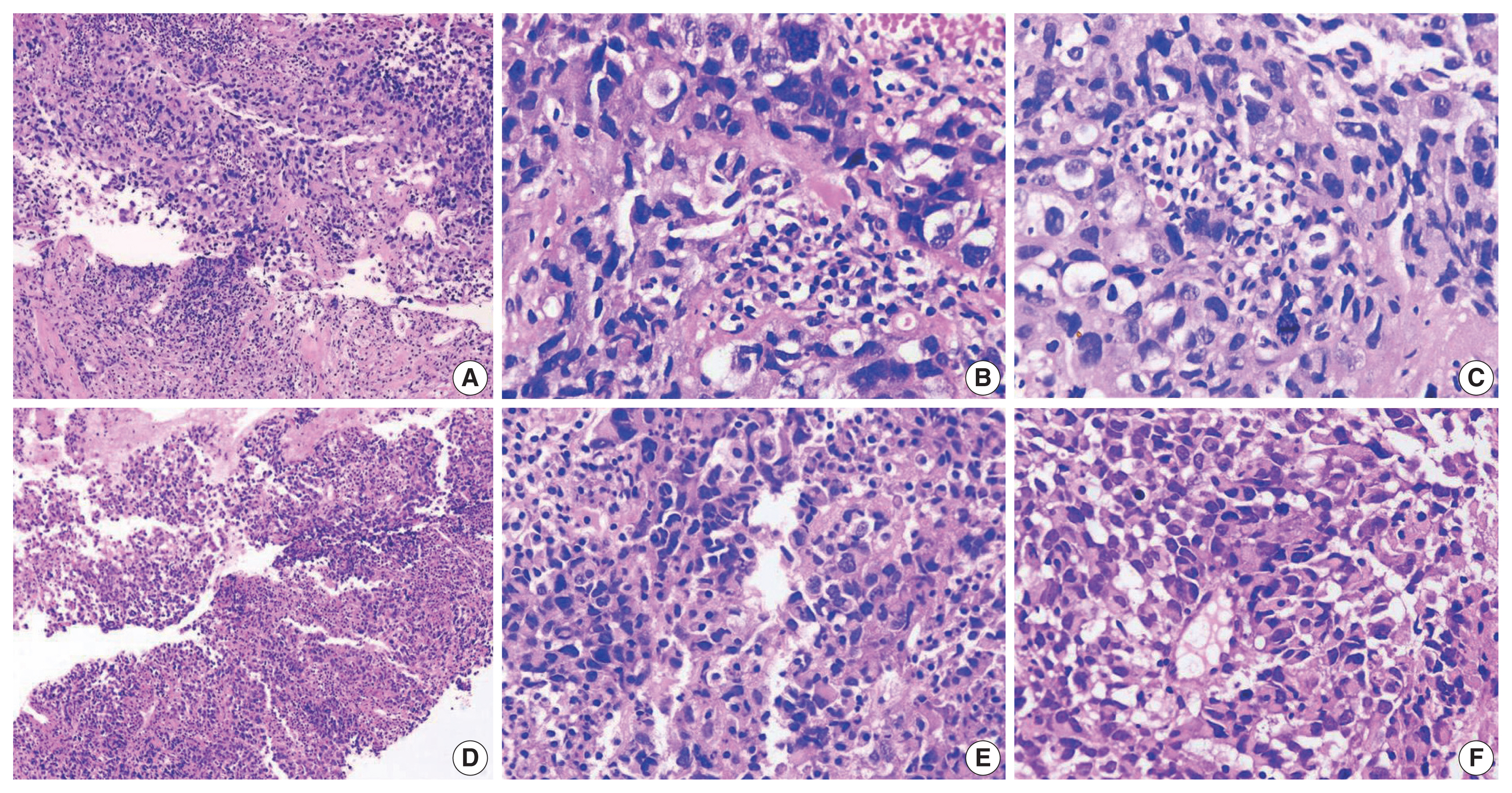
Histology images of cases. (A–C) Case No. 8 of SMARCA4/BRG1 protein–deficient thoracic sarcoma (SD-TS) showed a diffuse growth pattern. The constituent cells are large, and some have clear cytoplasm. Nuclei are irregular. (D, E) The stroma is inflamed in SD-TS. (E, F) Two cases (cases No. 7 and 9) revealed tumors with diffuse growth but obvious rhabdoid morphology of spheroidal cytoplasm, eccentric nuclei, and globoid inclusions.
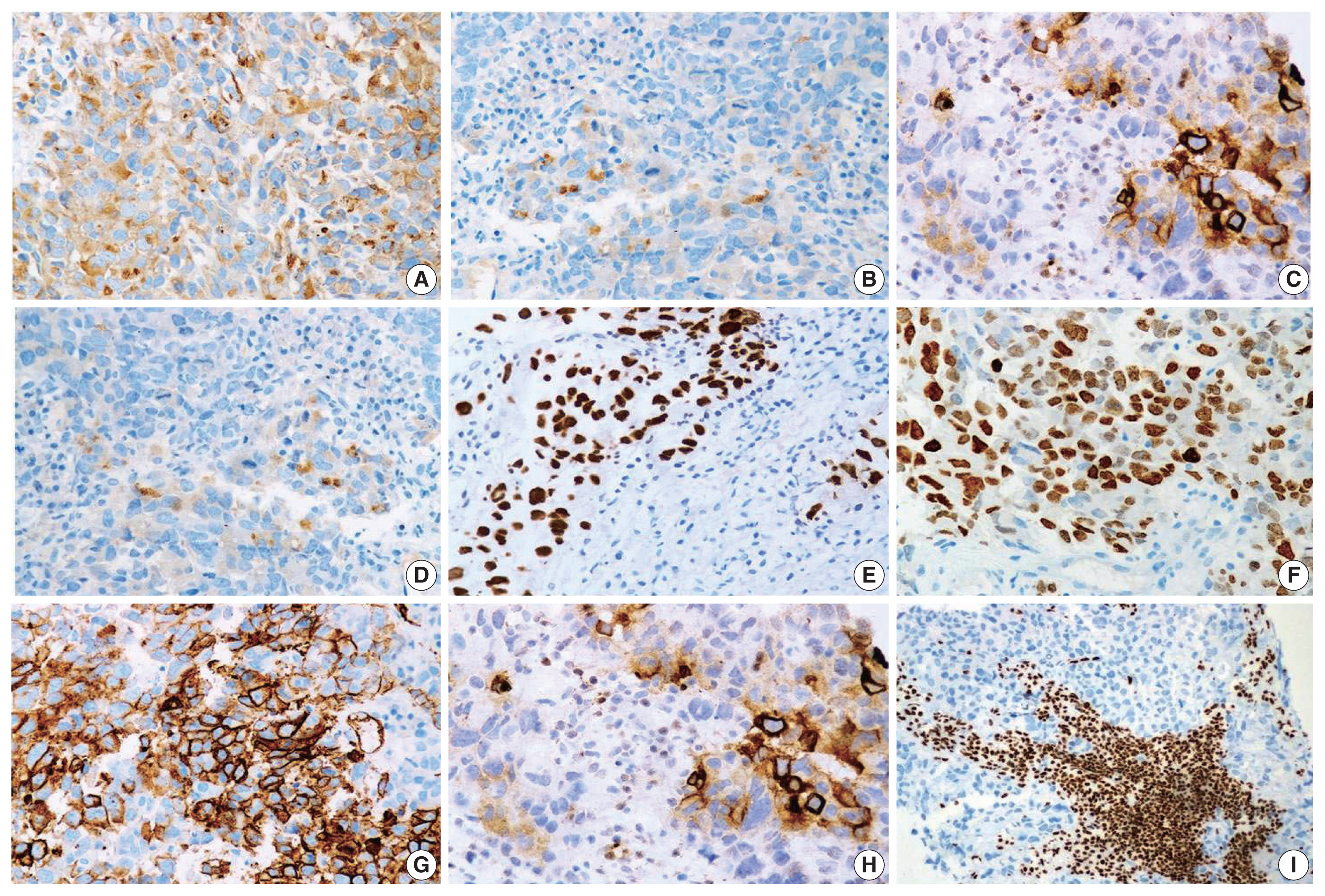
Immunophenotype of SMARCA4/BRG1 protein–deficient thoracic sarcoma (SD-TS). (A) Weak expression of pan-cytokeratin (CK) was observed in all cases of SD-TS. (B) Weak CK7 immunoreactivity was noted in case No. 7. (C) Focal strong CK7 staining is seen in case No. 8. (D) Case No. 7 expressed weak and focal Hep Par 1. (E–G) Expression of stemness markers of SALL4, SOX2, and CD34, respectively. (H) Expression of synaptophysin in case No. 7 of SD-TS. (I) Lack of nuclear expression of BRG1 defining BRG1 loss. Note the intense staining of stromal and inflammatory cells.
Molecular analysis
On molecular analysis, no actionable mutation in EGFR, ALK-1, or ROS1 gene was identified in SD-TT (Table 3). The targeted panel for biomarker detection in case No. 1 showed a copy number gain of c-MYC gene to 7.46 against the normal ploidy of 2, which was confirmed by FISH (copy number gain of 8.2 signals per cell). Significant PD-L1 (≥ 1%) expression was seen in two cases (case Nos. 4 and 6) (Table 3).
Clinical outcome
Follow-up information was available for all cases of SD-LUAD (Table 1). Three cases of SD-LUAD (case Nos. 1, 2, and 5) were on supportive care and died within two weeks of diagnosis. One case (case No. 4), which was reclassified as SD-LUAD and had received multiple lines of chemotherapy, died 23 months after initial diagnosis and within one month of repeat biopsy. All cases of SD-TS opted for further treatment in their respective cities and are alive, as learned telephonically. The duration of observation in this subgroup was 3–4 months, but the status of objective response and nature of therapy are not known.
DISCUSSION
Mammalian SWI/SNF chromatin remodeling is dependent on ATPase activity that resides in BRG1 or its ortholog brahma [1,3,4]. Two thoracic tumors are caused by somatic loss of heterozygosity resulting from biallelic loss of SMARCA4, namely SD-LUAD and SD-TS. The existing literature on these tumors has brought greater awareness about clinicopathologic characteristics, prognosis, and therapeutic consequences [10–17].
In a recently published, large series on SD-TT, the majority of cases was male, chronic smokers, and ranged in age from 30–80 years (mean, 58 years). Most had large thoracic mass with bulky lymphadenopathy, and nearly all patients had stage IV disease with bone metastasis [17]. Seven of our nine cases of SD-TT were also male, middle aged to elderly, and chronic smokers. Similarly, all our SD-LUAD cases had significant lung mass and stage IV disease with bony metastasis. A surprising observation in our cohort was lack of bone metastasis in SD-TS, whereas all cases of SD-LUAD had extensive bone involvement. This observation contradicts the dedifferentiation hypothesis [14,16] for emergence of SD-TS from SD-LUAD, though weak expression of epithelial markers in SD-TS and stem cell markers in SD-LUAD suggest the possibility of transition from SD-LUAD to SD-TS.
Morphological patterns of SD-LUAD have been recognized, from less common, well-differentiated to more common, poorly-differentiated malignant tumors [11]. All our cases had a predominant solid growth pattern with scattered clear to signet ring cells and inflamed stroma, similar to the findings seen in other studies [11,12]. Scattered cells with intracytoplasmic mucin were noted in four of six cases. This observation replicates the findings of Agaimy et al. [11]. Two cases with trabecular pattern of TTF1 negativity and Hep Par 1 expression, could easily be misinterpreted as hepatoid adenocarcinomas or hepatocellular carcinomas without BRG1 testing. We contend that all cases of malignancy of unknown origin (MUO) with hepatoid immunophenotype be tested for lack of BRG1 expression to correctly diagnose SD-TT. However, the aberrant expression of Hep Par 1 has not been explained well in the existing literature and is possibly a result of extensive chromatin remodeling associated with SMARCA4 loss. SD-LUAD tumors were negative for p40, TTF1, Napsin A, neuroendocrine markers, and CK5/6 but expressed CK7 in four of the six cases and Hep Par 1 and EMA in all cases, which was in line with observation from other studies [10–12]. Two cases of SD-LUAD (case Nos. 3 and 5) with scattered rhabdoid cells, despite showing diffuse positivity for CK, were negative for CK7 and had weak and focal Hep Par 1 expression and weak SOX2 and CD34, which placed them between SD-LUAD and SD-TS. These cases represent a possible transition toward SD-TS and support the concept of a biologic continuum between these tumors [17,19]. Notably, CK and EMA expression levels were strong in these two cases, unlike the other SD-TS, allowing us to categorize them as SD-LUAD.
The prevailing literature remains controversial as to whether SD-TS represent a distinct entity and, if so, whether there is an evolutionary relation between SD-LUAD and SD-TS [14,16,17]. There are no unambiguous clinico-radiological or histopathological findings to differentiate between them [10–17]. Perret et al. [14] proposed criteria for SD-TS of 1) rhabdoid or poorly differentiated phenotype; (2) complete loss of expression of SMARCA4 and SMARCA2; and (3) focal or diffuse expression of at least two of the following markers: SOX2, CD34, or SALL4.
SD-TS patients in our case series had dominance of rhabdoid tumor cells, but the overall morphology was indistinguishable from that of SD-LUAD. No spindle cell cytology, myxoid alterations, or a known growth pattern exclusive to sarcomatous histogenesis was observed. Separation from the latter was achieved solely by immunophenotyping, which revealed diffuse and strong staining for stem cell markers CD34, SOX2, and SALL4 and focal staining for keratin and Hep Par 1 in SD-TS in two of the three cases. Also, these two cases had no expression of EMA. The third case had undifferentiated round cell to rhabdoid morphology with BRG1 loss but focal staining for CD34 and CK and weak, focal staining for EMA. In such cases, other tumors like epithelioid mesotheliomas, which can show BRG1 loss, must be excluded by clinico-radiological features and negativity for other mesothelial markers (CK5/6, calretinin, and WT1) [14]. Further, complete absence or weak focal presence of EMA with focal positivity for CK helps to exclude sarcomatoid/undifferentiated carcinomas.
Predictive biomarker testing for sensitizing EGFR mutation and ALK-1 and ROS1 rearrangement was negative in all SD-TT cases in this cohort. Case No. 1, which was tested by massively parallel sequencing for broad predictive biomarkers, showed copy number gain for c-MYC but none of the actionable genetic alterations. Lack of currently druggable genetic alterations is the hallmark of SD-LUAD. This finding in our series is a reiteration of similar findings in previous studies [19,21]. This confirms the futility of expensive biomarker testing in this subset of lung adenocarcinoma and highlights the need to filter such cases upfront to avoid wasting effort and resources. Furthermore, for these tumors with a different biology and an aggressive course with no actionable drivers yet, a better understanding of their mechanistic nuances with new and efficacious therapeutic options are needed [22], some of which such as EZH2 inhibitors [23] and immune check point inhibitors [24,25] have started emerging. To fulfill the referred objective of differentiating SD-TT from NSCC-NOS, we followed a simple tissue proficient diagnostic schema as shown in Fig. 5.
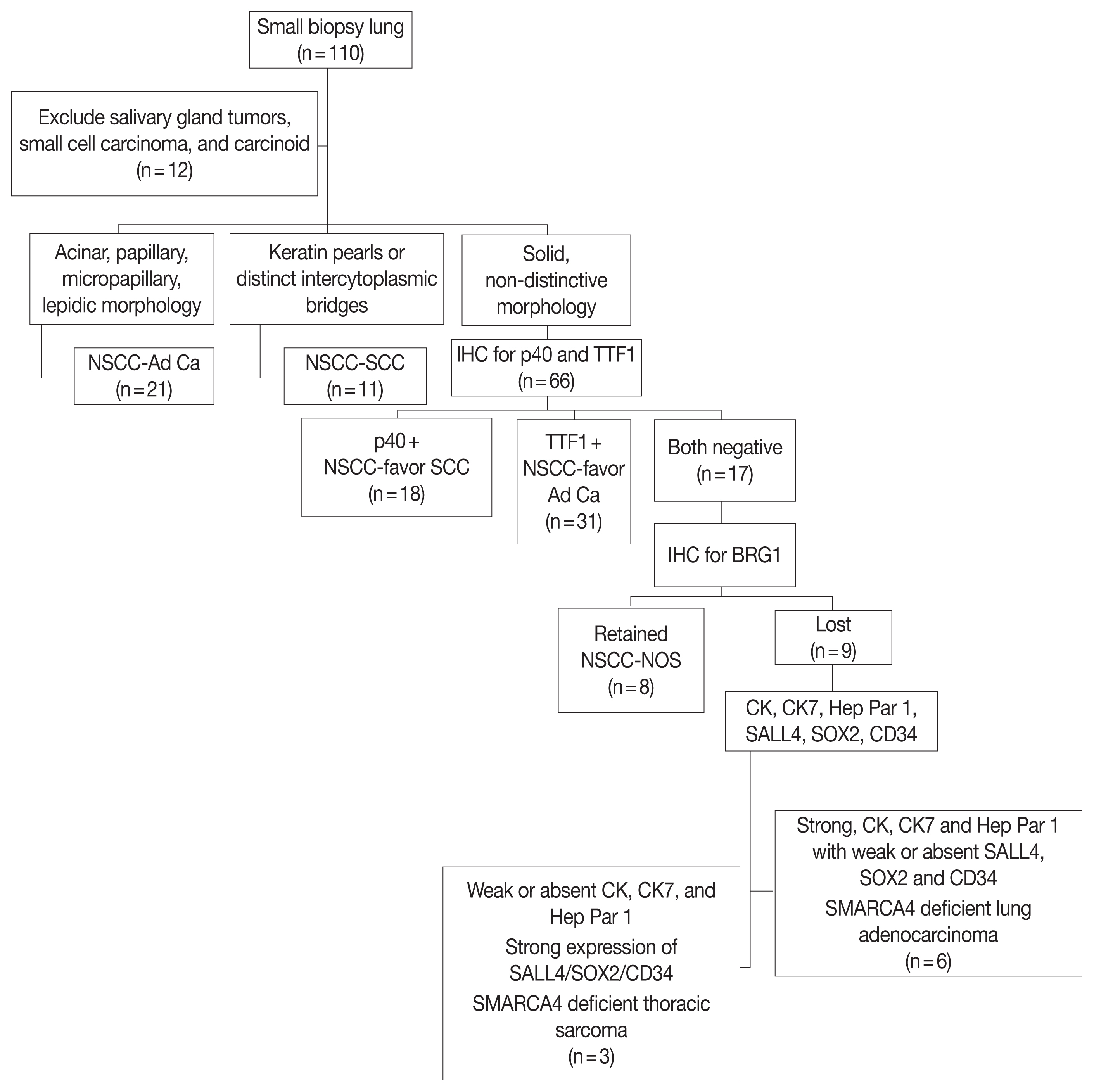
Diagnostic schema for SMARCA4/BRG1 protein-deficient thoracic tumors. NSCC-Ad Ca, non-small cell carcinoma adenocarcinoma; SCC, squamous cell carcinoma; IHC, immunohistochemistry; TTF1, transcription termination factor 1; CK, cytokeratin.
There are certain limitations in the present study. First, the number of cases studied is small. Second, the study only confirmed that BRG1 loss is confined to TTF1-negative NSCLC based on small tissue microarray and might not agree with all existing literature [10]. Third, no confirmatory molecular testing targeting SMARCA4 gene mutation was performed. Fourth, the diagnostic pathway requires extensive use of IHC to separate SD-TT into SD-LUAD and SD-TS once BRG1 loss is established. However, such extension of IHC is acceptable in the BRG1-deficient subset that lacks actionable biomarkers and omits biomarker testing without therapeutic impact.
To conclude, SD-LUAD and SD-TS are difficult to differentiate from each other and from other NSCLC based on morphology alone. They are likely to be reported as NSCC-NOS with immunonegativity to p40 and TTF1, necessitating biomarker testing with a waste of time and resources. We argue that the diagnostic pathway presented here can help to diagnose such cases so that they can be studied more effectively for new biomarkers and therapeutic techniques. Overall, this study highlights the need and method of delineating SD-TT in reporting of small biopsy for NSCLC and shares a useable workflow to clarify tumor type. Finally, awareness of this entity can help prevent misdiagnosis of MUO as hepatic or hepatoid carcinomas with consequent negative effects.
Notes
Ethics Statement
All procedures performed in the current study were approved by Institutional Review Board (vide letter no: RGCIRC/IRB-BHR/33/2021 dated 6th January 2021) in accordance with the 1964 Helsinki declaration and its later amendments. Informed consent was obtained from all individual participants included in the study.
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: AM. Data curation: DB, RT. Formal analysis: AM, DB. Investigation: AM, DB, AJ. Methodology: AM, DB. Resources: RT, AJ. Supervision: AM. Visualization: AM, DB. Writing—original draft: DB, AM, RT. Writing—review & editing: AM, DB. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.