Clinicopathologic implication of PD-L1 gene alteration in primary adrenal diffuse large B cell lymphoma
Article information

Abstract
Background
Primary adrenal (PA) diffuse large B cell lymphoma (DLBCL) was previously reported as an aggressive subset of DLBCL, but its genetic features were not sufficiently characterized. From our previous study of DLBCL with programmed death-ligand 1 (PD-L1) gene alterations, we focused on PD-L1 gene alterations in PA-DLBCL with clinicopathologic implications.
Methods
We performed fluorescence in situ hybridization for PD-L1 gene translocation and amplification in PA-DLBCL (n = 18) and comparatively analyzed clinicopathologic characteristics with systemic non-adrenal (NA)-DLBCL (n = 90).
Results
PA-DLBCL harbored distinctive features (vs. NA-DLBCL), including high international prognostic index score (3–5) (72% [13/18] vs. 38% [34/90], p = .007), poor Eastern Cooperative Oncology Group performance score (≥ 2) (47% [7/15] vs. 11% [10/90], p = .003), elevated serum lactate dehydrogenase (LDH) (78% [14/18] vs. 51% [44/87], p = .035) and MUM1 expression (87% [13/15] vs. 60% [54/90], p = .047). Moreover, PA-DLBCL showed frequent PD-L1 gene alterations (vs. NA-DLBCL) (39% [7/18] vs. 6% [5/86], p = .001), including translocation (22% [4/18] vs. 3% [3/87], p = .016) and amplification (17% [3/18] vs. 2% [2/87], p = .034). Within the PA-DLBCL group, PD-L1 gene–altered cases (vs. non-altered cases) tended to have B symptoms (p = .145) and elevated LDH (p = .119) but less frequent bulky disease (≥ 10 cm) (p = .119). In the survival analysis, PA-DLBCL had a poor prognosis for overall survival (OS) and progression-free survival (PFS) (vs. NA-DLBCL; p = .014 and p = .004). Within the PA-DLBCL group, PD-L1 translocation was associated with shorter OS and PFS (p < .001 and p = .012).
Conclusions
PA-DLBCL is a clinically aggressive and distinct subset of DLBCL with frequent PD-L1 gene alterations. PD-L1 gene translocation was associated with poor prognosis in PA-DLBCL.
Malignant lymphomas have been classified and updated by distinctive morphologic, clinical, and genetic features [1]. Classical Hodgkin lymphoma (cHL) has activated programmed death-ligand 1 (PD-L1) signaling, while it is derived from B cells [2–5]. For B cell-derived neoplasms with intact B cell antigen expression, including diffuse large B cell lymphoma (DLBCL), the clinicopathologic significance of PD-L1 gene alteration has not been well characterized. In our previous retrospective study of DLBCL, PD-L1 gene alterations, including translocation and amplification, were observed in 14% of non-germinal center B cell-like (non-GCB) subtype [6]. Interestingly, frequent alteration of the PD-L1 gene was observed in several anatomic site-specific subtypes of DLBCL, including primary mediastinal large B cell lymphoma, primary DLBCL of the central nervous system (CNS) and primary testicular DLBCL [7,8]. These phenomena suggest that PD-L1 gene alteration may also play an important pathogenic role in a delicately defined subset of DLBCL.
Adrenal gland has a unique microenvironment with potential features of immune sanctuary sites and locally increased immunosuppressive hormones [9,10]. It is rarely involved by malignant lymphomas usually with secondary spread from systemic extra-adrenal lymphomas [11]. Primary adrenal lymphoma accounts for only < 1% of non-Hodgkin lymphomas and 3% of extranodal lymphomas, and DLBCL is the most common histologic type [11,12]. Primary adrenal DLBCL (PA-DLBCL) was reported to frequently have clinicopathologic features such as elevated lactate dehydrogenase (LDH), presence of B symptoms, non-GCB subtype, Bcl-6 gene rearrangement and worse prognosis [11,13,14].
In our previous study of DLBCL, PD-L1 gene alteration was observed in one out of three cases of the PA-DLBCL subset, providing a clue for further investigation of PD-L1 gene alteration in PA-DLBCL [6]. Furthermore, we also focused on several studies relating PD-L1 upregulation with clinical aggressiveness [15,16]. These findings raised questions about the prevalence of PD-L1 gene alterations in PA-DLBCL as well as its role in presumed clinical aggressiveness [14]. Herein, we hypothesized that the clinicopathologic features of PA-DLBCL may be associated with PD-L1 gene alterations. We investigated (1) the clinicopathologic characteristics of PA-DLBCL, including the frequency of PD-L1 gene alterations, and (2) the associations between PD-L1 gene alterations and clinicopathologic variables, including prognosis.
MATERIALS AND METHODS
Patients and samples
From 20 cases of DLBCL, not otherwise specified (NOS) that occurred primarily in the adrenal gland, i.e., “primary adrenal DLBCL (PA-DLBCL)” retrospectively retrieved from the archives of pathology records between May 2003 and January 2013 at Seoul National University Bundang Hospital (SNUBH) and Seoul National University Hospital, two cases were excluded due to inappropriate tissue conditions and failure to meet the clinical criteria. Therefore, a total of 18 cases of PA-DLBCL were finally enrolled. To elucidate the unique clinicopathologic features of PA-DLBCL, systemic cases of DLBCL, NOS diagnosed at SNUBH during the same period was used as a reference group of “non-adrenal (NA)-DLBCL” group containing 90 cases [6]. Considering the characteristics of the previously described PA-DLBCL [11,13], we defined PA-DLBCL as “de novo DLBCL with one or more adrenal gland mass-forming and/or hypermetabolic lesion(s) as the most dominant lesion” for the purpose of the present study. Specifically, the “PA-DLBCL” group included the following cases: (1) cases of DLBCL, NOS according to the World Health Organization (WHO) 2016 criteria with no prior history of lymphoma, (2) cases with unequivocal adrenal-dominant mass lesions (maximal diameter: adrenal mass > ×2 of NA mass) measured by computed tomography (CT) and/or 18F-positron emission tomography (PET)-CT, and (3) cases proven by appropriate pathologic studies. The following cases were excluded from the “PA-DLBCL” group for the purpose of clarity of this study: (1) widely disseminated DLBCL cases, (2) cases with widespread node-based disease, and (3) any distinct entities including ‘DLBCL of the CNS’, ‘primary mediastinal large B cell lymphoma’, ‘primary testicular DLBCL’, so-called ‘gray-zone lymphomas’ and lymphomas associated with Epstein-Barr virus or human immunodeficiency virus. All samples in this study were archived materials of formalin-fixed paraffin-embedded tissues obtained from biopsy or surgical specimens.
The pathologic diagnosis was reviewed by two pathologists (KRL and JHP) by WHO 2016 criteria, and the ‘cell of origin’ subtype was determined by Han’s algorithm [1,17]. Clinical findings were retrieved from medical records.
Fluorescence in situ hybridization for PD-L1 gene
We investigated the genetic alterations of the PD-L1 (CD274) gene (translocation, copy number gain, and amplification) by using fluorescence in situ hybridization (FISH) staining, as previously described [6]. A PD-L1 break-apart probe (9p24.1) (catalog No. PDL1BA-20-ORGR) from Empire Genomics (Buffalo, NY) was used to detect the translocation, and a PD-L1 (orange)/chromosome 9 (green) probe set (9p24.1/9p21.33) (catalog No. PDL1-CHR09-20-ORGR) from the same company was used to detect the copy number gain and amplification. To determine translocation, copy number gain and amplification, more than 200 cells with non-overlapping nuclei were counted. Separation of orange and green signals in more than 15% of cells was interpreted as the presence of translocation. Copy number gain and amplification were defined as orange/green signal (PD-L1 gene/chromosome 9) ratios > 2 and > 4, respectively. In the case of clustered signals, we counted them as 12 copies. In our series of PA-DLBCL, there were no cases with copy number gain or cases with concurrent translocation and copy number gain/amplification.
Statistical analysis
SPSS Statistics ver. 19 (IBM Corp., Armonk, NY, USA) was used for statistical analysis. Chi-square and Fisher exact tests were used to compare clinicopathologic features. Kaplan-Meier analysis was used to draw survival curves using the log-rank test. A Cox proportional hazard model was used for multivariate survival analysis. Overall survival (OS) was defined as the time interval from the date of diagnosis to the date of last follow-up or death. Progression-free survival (PFS) was defined as the time interval from the date of treatment to the date of progression with radiologic confirmation using CT and/or PET-CT after treatment or the date of death. All p-values reported were two-sided, and statistical significance was accepted when they were less than .05.
RESULTS
Clinicopathologic characteristics of PA-DLBCL
To characterize the clinicopathologic features of PA-DLBCL, we analyzed the clinicopathologic parameters of the PA-DLBCL group with a reference group of NA-DLBCL (Table 1). As for clinical variables, compared to NA-DLBCL, PA-DLBCL patients showed high international prognostic index (IPI) score (3–5) (72% [13/18] in PA-DLBCL vs. 38% [34/90] in NA-DLBCL, p = .007), poor Eastern Cooperative Oncology Group performance score (ECOG PS) (≥ 2) (47% [7/15] in PA-DLBCL vs. 11% [10/90] in NA-DLBCL, p = .003), elevated LDH (78% [14/18] in PA-DLBCL vs. 51% [44/87] in NA-DLBCL, p = .035) with trends for high frequency of B symptoms (44% [8/18] in PA-DLBCL vs. 23% [21/90] in NA-adrenal DLBCL, p = .082) and bulky disease (≥ 10 cm) (22% [4/18] in PA-DLBCL vs. 8% [7/90] in NA-adrenal DLBCL, p = .084). However, the two groups showed a similar distribution of high-stage disease (50% [9/18] in PA-DLBCL vs. 53% [48/90] in NA-DLBCL).

Clinicopathologic features of PA-DLBCL and NA-DLBCL patients
Pathologically, the PA-DLBCL group showed frequent MUM1 expression (87% [13/15] in PA-DLBCL vs. 60% [54/90] in NA-adrenal DLBCL, p = .047) with trends for Bcl-6 expression (73% [11/15] in PA-DLBCL vs. 49% [44/90] in NA-adrenal DLBCL, p = .079).
Taken together, PA-DLBCL group harbored aggressive clinicopathologic features even with similar stage, compared to NA-DLBCL.
PD-L1 gene alteration by using fluorescence in situ hybridization
We next analyzed the frequency of PD-L1 gene alterations, including translocation and amplification, by using the FISH method. As shown in Table 2 and Fig. 1, PD-L1 gene alteration in PA-DLBCL accounted for 39% (7/18), including translocation in 22% (4/18) and amplifications in 17% (3/18), which were much higher frequencies than NA-DLBCL with PD-L1 gene alteration in 6% [5/86], including translocation in 3% (3/87) and amplification in 2% (2/87) (p = .001, p = .016, and p = .034, respectively). Therefore, the PA-DLBCL group had a significantly higher frequency of PD-L1 gene alterations than the NA-DLBCL group.

PD-L1 genetic alteration in PA-DLBCL and NA-DLBCL
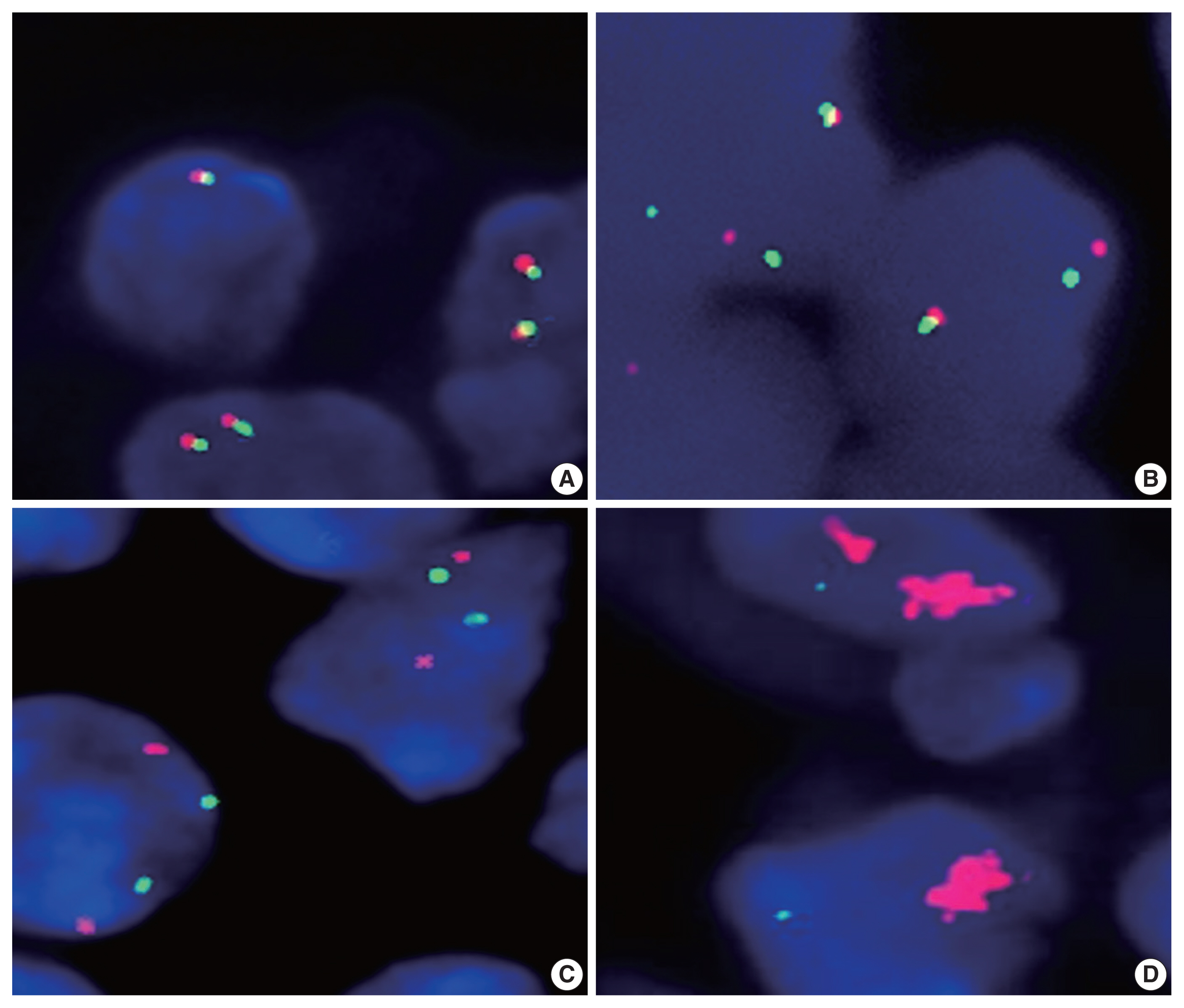
Fluorescence in situ hybridization analysis of the programmed death-ligand 1 (PD-L1) gene in diffuse large B cell lymphoma. By using a dual-color orange/green break-apart probe, non-split fused signal indicates the absence of translocation (A), while separate orange and green signals indicate the presence of translocation (B). By using a copy number analysis probe, orange (PD-L1 gene) and green (chromosome 9) signals showed a nearly 1:1 ratio in cases with no gain or amplification of the PD-L1 gene (C), in contrast to amplified orange signals indicating PD-L1 gene amplification (D).
Associations between clinicopathologic variables and PD-L1 gene alterations in PA-DLBCL
We next investigated associations between PD-L1 gene alterations and clinicopathologic parameters within the PA-DLBCL group. As shown in Table 3, PA-DLBCL with PD-L1 gene alteration tended to have elevated serum LDH (100% [7/7] in the PD-L1–altered group vs. 64% [7/11] in non-altered group) and B symptoms (71% [5/7] in the PD-L1–altered group vs. 27% [3/11] in non-altered group). In contrast, there was a tendency for less frequent bulky disease (≥ 10 cm) in the PD-L1–altered group (0% [0/7] in the PD-L1–altered group vs. 36% [4/11] in non-altered group).
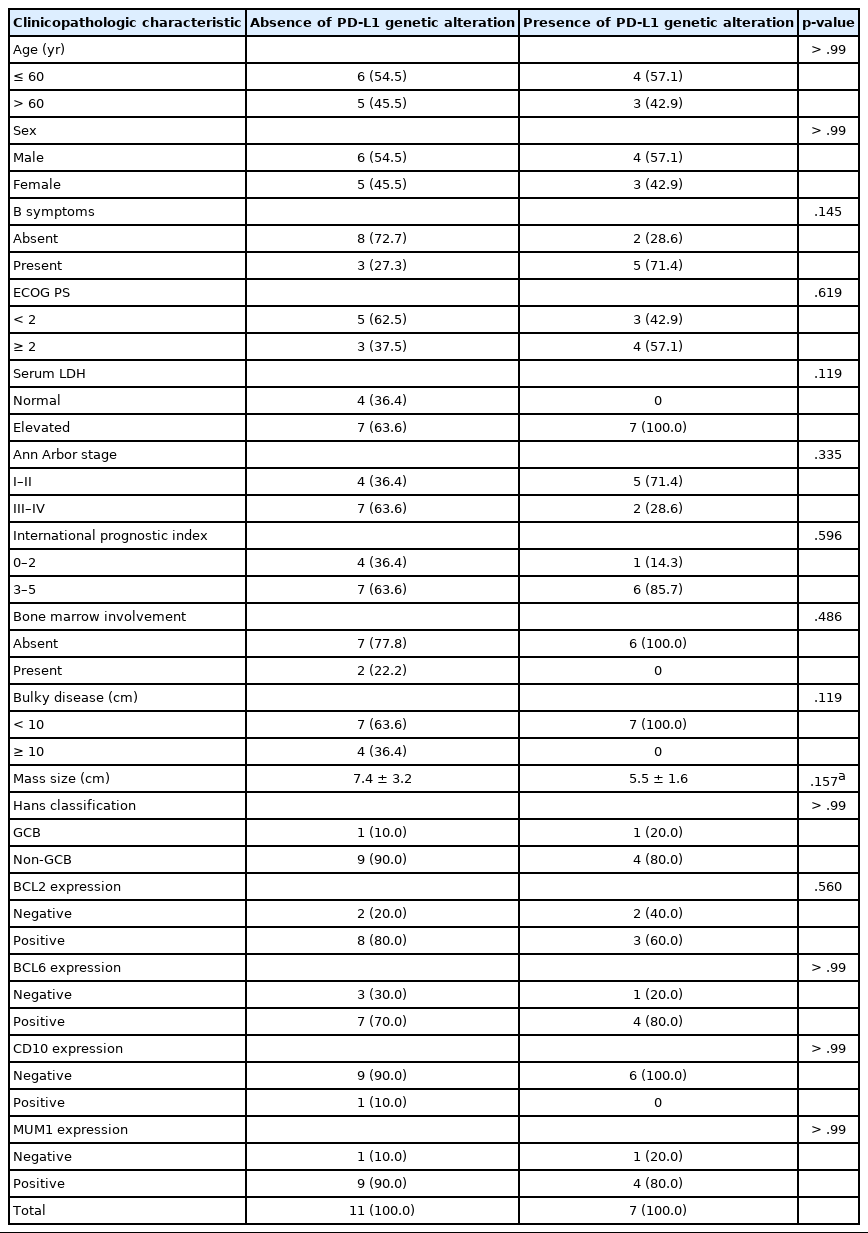
Clinicopathologic features of primary adrenal diffuse large B cell lymphoma according to PD-L1 gene alteration
Survival analysis
As shown in Fig. 2, the PA-DLBCL group showed inferior OS and PFS compared to the NA-DLBCL group (p = .014 and p = .004) (Fig. 2A, B). When survival was analyzed within the PA-DLBCL group (Table 4), PD-L1 gene alteration did not significantly affect OS or PFS (p > .05) (Fig. 2C, D). When the survival effect of PD-L1 translocation was analyzed, the PD-L1 translocation group showed inferior OS and PFS (p < .001 and p = .012) (Fig. 2E, F). However, PD-L1 gene amplification was not associated with prognosis. Among the conventional prognostic markers, ECOG PS was a significant predictor for PFS (p = .039). In multivariate analysis, all variables were not significant due to the limited number of cases.
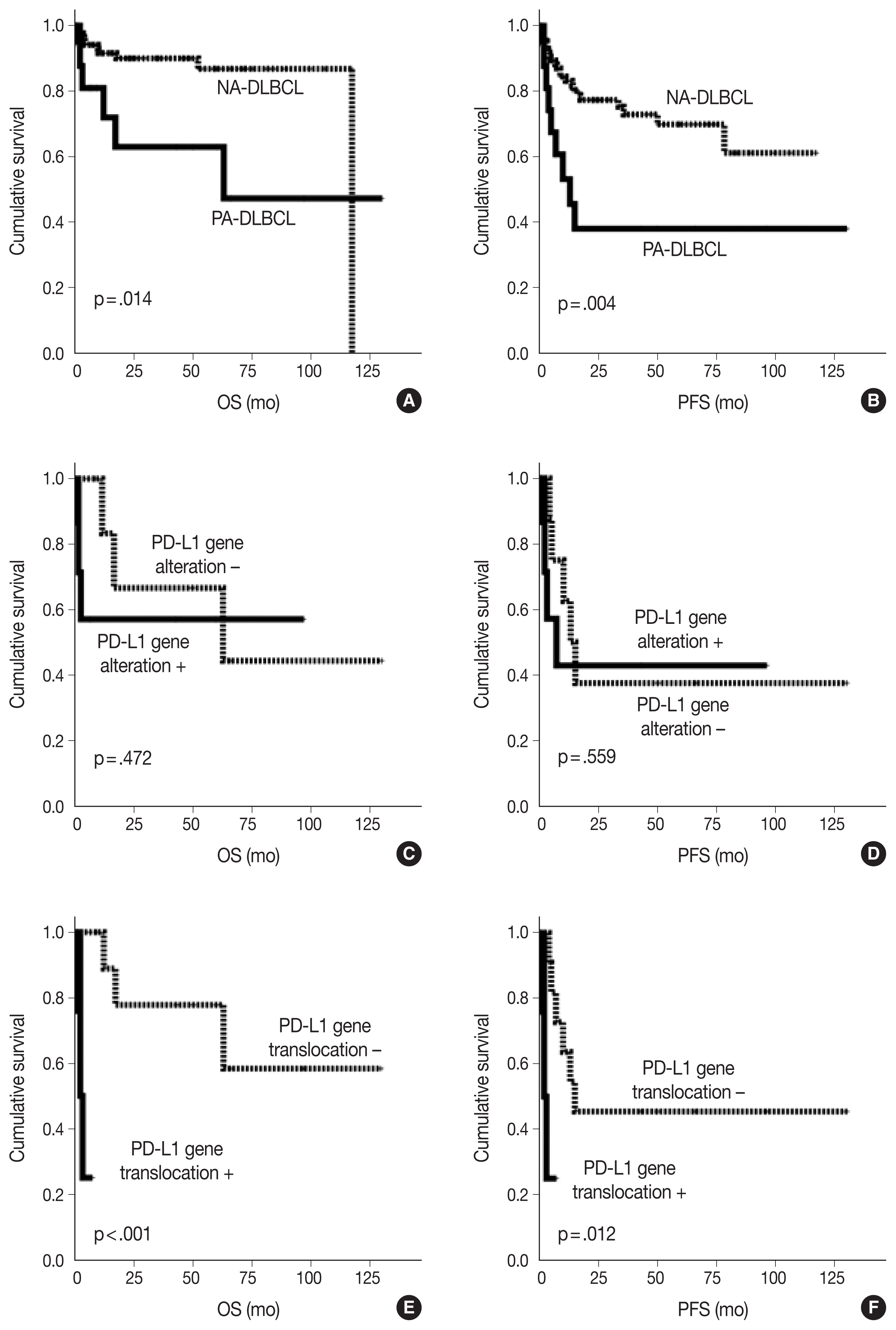
Kaplan-Meier curves of overall survival (OS) and progression-free survival (PFS) in diffuse large B cell lymphoma (DLBCL) patients. OS (A) and PFS (B) of primary adrenal (PA)- and non-adrenal (NA)-DLBCL patients. OS (C) and PFS (D) of PA-DLBCL patients by the presence of programmed death-ligand 1 (PD-L1) gene alterations. OS (E) and PFS (F) of PA-DLBCL patients by the presence of PD-L1 gene translocation.
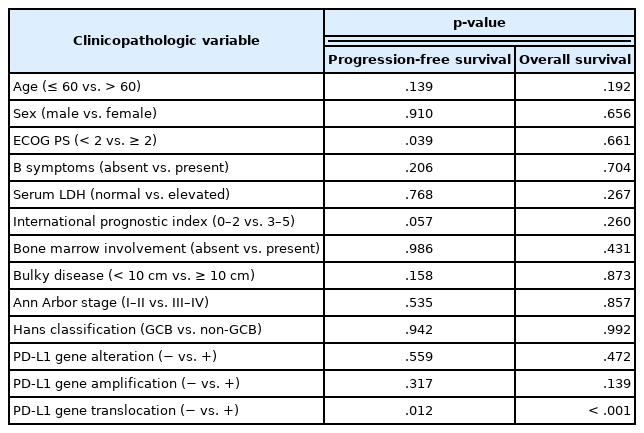
Univariate survival analysis according to clinicopathologic variables and PD-L1 gene alteration in primary adrenal diffuse large B cell lymphoma
DISCUSSION
In our study, we demonstrated that PD-L1 gene alterations were more frequent in PA-DLBCL than in NA-DLBCL and tended to be associated with aggressive clinical parameters such as the presence of B symptoms or high LDH. Patients with PD-L1 translocation but not amplification revealed inferior survival in PA-DLBCL.
DLBCLs in certain primary anatomic sites, such as the mediastinum, CNS or testis, are classified as specific types because of their distinct molecular pathogenesis. PD-L1/PD-L2 signaling has been reported to be activated in these lymphomas [7,8]. Although PA-DLBCL is not defined as a special type based on anatomic sites, it is usually associated with aggressive features and distinct pathologic characteristics [12,14]. Our study also supported that PA-DLBCL had aggressive features with a high rate of PD-L1 gene alteration, suggesting unique clinicopathologic characteristics.
Because of the rarity of PA-DLBCL, its clinicopathologic features, including pathogenic mechanism and clinical behavior, have not been well characterized. Several previous reports revealed aggressive features of PA-DLBCL, such as frequent B symptoms, elevated serum LDH, bulky mass formation and predominance of the non-GCB subtype, which are in concordance with our results [11–14]. In the present study, compared to the NA-DLBCL group, the PA-DLBCL group showed frequent poor ECOG PS (≥ 2), high IPI (3–5) and poor prognosis, suggesting the intrinsic aggressive features of PA-DLBCL [11,12], even with a nearly equal proportion of high-stage (III–IV) disease in both groups (50% vs. 53%).
The biological role of the programmed death-1/PD-L1 axis was revealed as the induction and maintenance of peripheral tolerance in T cells to prevent autoimmune reactions. However, inhibitory signals via this pathway hamper the proliferation and function of effector T cells, resulting in immune evasion in antitumor immunity [18]. Recently, several studies have suggested that cancer cell-intrinsic PD-L1 signaling contributes to the proliferation, migration and invasion of various types of cancer cells, including lymphoma cells [19]. In this context, these extrinsic and intrinsic effects of PD-L1 signaling may help to constitute the features of PD-L1–altered cancers. In our PA-DLBCL group, we observed that PD-L1 gene alteration had trends for frequent B symptoms, elevated serum LDH and relatively less bulky mass lesions, suggesting that PD-L1–altered tumor cells may tend to secrete mediators producing systemic reactions at a relatively early phase of mass formation.
The aggressive clinicopathologic nature of PA-DLBCL could not be fully explained by PD-L1 gene alteration due to its limited prevalence (39%). Another genetic feature of PA-DLBCL includes Bcl-6 gene rearrangement [14], which is common in both GCB and non-GCB DLBCLs [1,20,21]. A recent functional study in a mouse model revealed that Bcl-6 suppressed PD-L1/PD-L2 expression in germinal center (GC) B cells [22]. However, the functional role of Bcl-6 on PD-L1 in the post-GC B cell context is not definite and remains to be investigated further.
In summary, we observed that PA-DLBCL had characteristic clinicopathologic features, including high IPI, inferior outcome, and frequent PD-L1 gene alteration. Within this PA-DLBCL group, PD-L1 gene translocation was a significant poor prognostic marker predicting OS and PFS. Our results suggest that PA-DLBCL can be a distinct entity with frequent PD-L1 genetic alterations and may provide insight into the potentially shared pathogenesis of DLBCL of the mediastinum, CNS, testis, and adrenal gland.
Notes
Ethics Statement
This study was approved by the Institutional Review Board of Seoul National University Bundang Hospital (B-1306-208-301) and Seoul National University (No. 1506-080-681), and informed consent was waived due to the retrospective design using archived material in this study.
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: KRL, JHP. Data curation: KRL, JK, YKJ, HJK, JOL, JHP. Formal analysis: KRL, JHP. Funding acquisition: JHP. Investigation: KRL, JK, YKJ, JOL, HJK, JHP. Methodology: KRL, JHP. Writing—original draft: KRL, JHP. Writing—review & editing: KRL, JK, YKJ, JOL, JHP. Approval of final manuscript: all authors.
Conflicts of Interest
J.H.P., a contributing editor of the Journal of Pathology and Translational Medicine, was not involved in the editorial evaluation or decision to publish this article. All remaining authors have declared no conflicts of interest.
Funding Statement
This study was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (NRF-2019R1F1A1061920) and the Development Fund from Seoul National University (grant No.: 800-20190386) funded by the HUNKIM Family Charitable Foundation.