Hepatic carcinoma expressing inhibin: case report of a proposed novel entity and review of the literature
Article information

Abstract
Hepatic carcinoma expressing inhibin is a recently described neoplasm with varied architecture, including trabecular, pseudoglandular, follicular/microcystic, organoid, solid and tubular patterns of growth. We report a case of hepatic carcinoma expressing inhibin that occurred in a 47-year-old woman presenting with epigastric and back pain. The tumor was located in the left hepatic lobe and measured 12 cm in diameter. On immunohistochemical stains, the neoplastic cells were positive for inhibin, as well as cytokeratins 7, 8/18 and 19. There was mild focal expression of synaptophysin, and lack of expression of hepatocytic markers. The histogenesis of hepatic carcinoma expressing inhibin is presently uncertain. From a practical point of view, this neoplasm can potentially cause diagnostic pitfalls by simulating other primary or metastatic tumors, such as hepatocellular carcinoma, cholangiocarcinoma, neuroendocrine tumors, and follicular carcinoma of thyroid gland. Performing inhibin immunostain could assist in the differential diagnosis of liver tumors with unusual histologic features.
Hepatic carcinoma expressing inhibin (HCEI) is a recently described neoplasm of uncertain histogenesis that exhibits histologic features simulating hepatocellular, cholangiocellular, thyroid, and neuroendocrine tumors. In addition to our original report in 2005 [1], fourteen cases of HCEI have been reported since 2017 [2–6], indicating that this neoplasm may be more common than previously thought. In most cases, the correct diagnosis was made in consultation; the contributors’ diagnoses often were those of neuroendocrine tumors [2–5].
We report a new case of HCEI, review the literature on this proposed novel entity, and discuss its possible histogenesis.
CASE REPORT
Clinical summary
We have recently examined in consultation a left hepatic lobe tumor from a 47-year-old woman, who presented with epigastric and back pain of several months’ duration. Physical examination revealed a palpable mass in the left upper abdominal quadrant. Her past medical history was unremarkable. The patient did not smoke or drink alcohol, had no history of liver disease, and was not on any medication. There was no family history of liver disease. On magnetic resonance imaging (MRI) of the upper abdomen, a lobulated mass was seen, which occupied almost the entire left hepatic lobe. The tumor appeared inhomogeneously enhanced after intravenous contrast administration. The remaining liver parenchyma appeared normal. There were no other abnormal findings in the pancreas, spleen, adrenal glands and kidneys. Chest X-ray was normal. Liver function tests, hematology work-up, and serum levels of tumor markers (carcinoembryonic antigen, CA19-9, CA125, and α-fetoprotein) were within normal limits. The patient underwent a left hepatic lobectomy. On postoperative chest computed tomography scan, as well as upper and lower abdominal MRI, there were no additional lesions or residual tumor.
Pathologic findings
On gross examination, the tumor was relatively well-circumscribed, displayed a tan-yellow cut surface, and measured 12 cm in greatest dimension (Fig. 1A). On microscopic examination, it exhibited trabecular, follicular and pseudoglandular architecture (Fig. 1B). The tumor cells were columnar or ovoid in shape, with eosinophilic cytoplasm and roundish nuclei with inconspicuous nucleoli (Fig. 1C). Mild nuclear pleomorphism and rare mitotic figures were found. The follicular structures contained amorphous eosinophilic material with peripheral vacuolization, reminiscent of thyroid follicles (Fig. 1B). Occasional fibrous septa were observed (Fig. 1B). Regions of hemorrhage and cystic degeneration were present. No necrosis was found. At the tumor border, there was invasive growth, with tongues and groups of neoplastic cells extending into the adjacent hepatic parenchyma (Fig. 1D). The hepatic parenchyma was otherwise unremarkable. No evidence of biliary intraepithelial neoplasia was present. The surgical margins of resection were free of tumor.
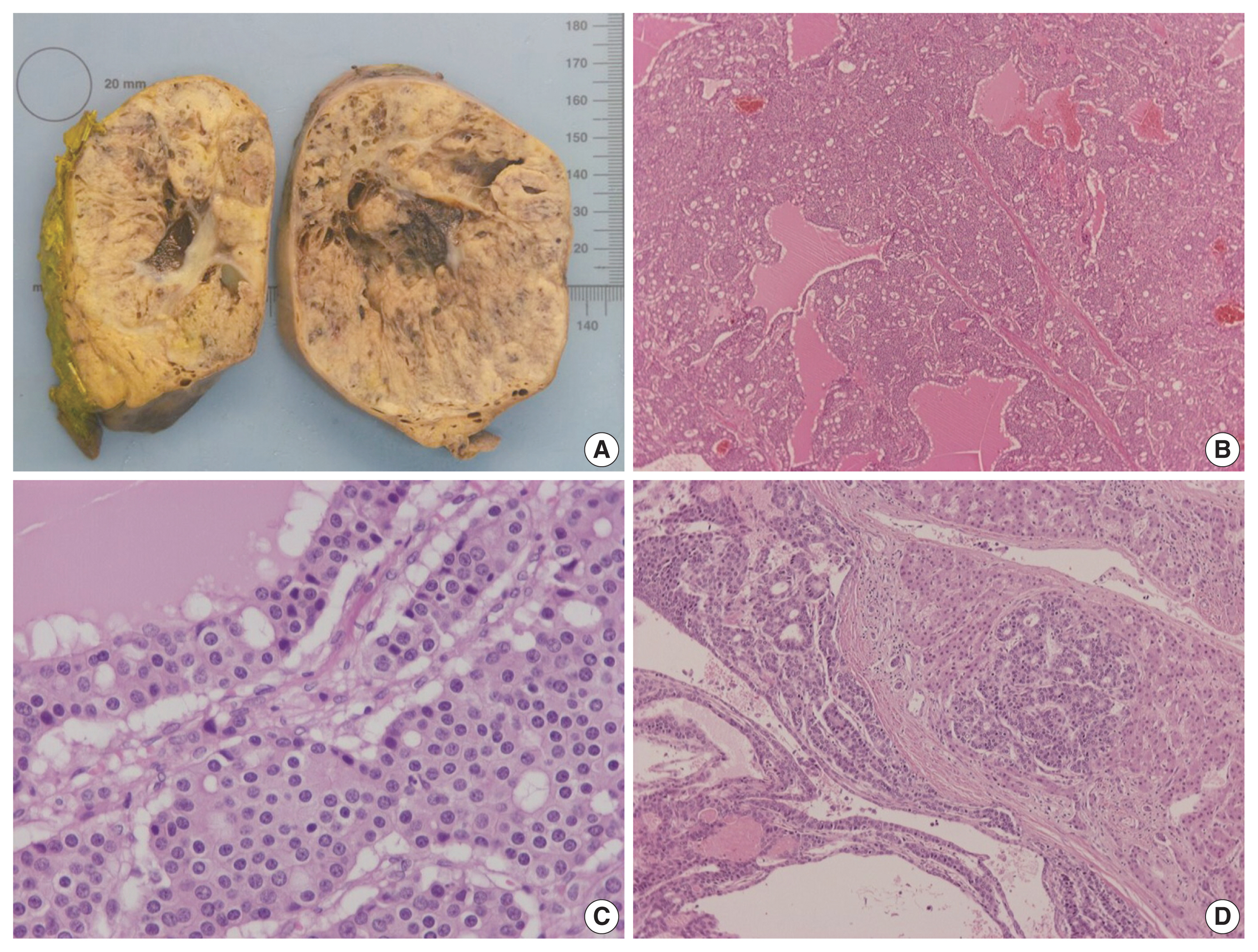
Gross and microscopic features of the tumor. (A) Gross examination shows a large, relatively well-circumscribed tumor, with a tan-yellow cut surface and focal areas of hemorrhage and cystic degeneration. (B) Representative low power view of the tumor, demonstrating trabecular, follicular and pseudoglandular architecture. (C) On high power examination, the neoplastic cells are columnar or ovoid, with eosinophilic cytoplasm and mild nuclear pleomorphism. (D) Neoplastic cell invasion of the adjacent hepatic parenchyma is evident at the tumor margin.
On immunohistochemical evaluation, the neoplastic cells were positive for cytokeratin (CK) 7 (Fig. 2A), CK8/18, and CK19 (Fig. 2B), as well as inhibin (Fig. 2C). There also was patchy mild positivity of tumor cells for synaptophysin (Fig. 2D). Membranous positivity with polyclonal carcinoembryonic antigen was present around pseudoglandular structures (Fig. 2E). The neoplastic cells were negative for HepPar-1, arginase-1, glypican-3, α-fetoprotein, glutamine synthetase, β-catenin (nuclear), CK20, epithelial membrane antigen, chromogranin, INSM1, thyroid transcription factor-1, thyroglobulin, human chorionic gonadotrophin, CA19-9, Melan A, CD10, CD56, CD99, vimentin, calretinin, estrogen and progesterone receptor proteins, PAX8, GATA3, CDX2, and trypsin. On immunohistochemical stains for Ki67 antigen, there was positivity in 10% of tumor cells (Fig. 2F).
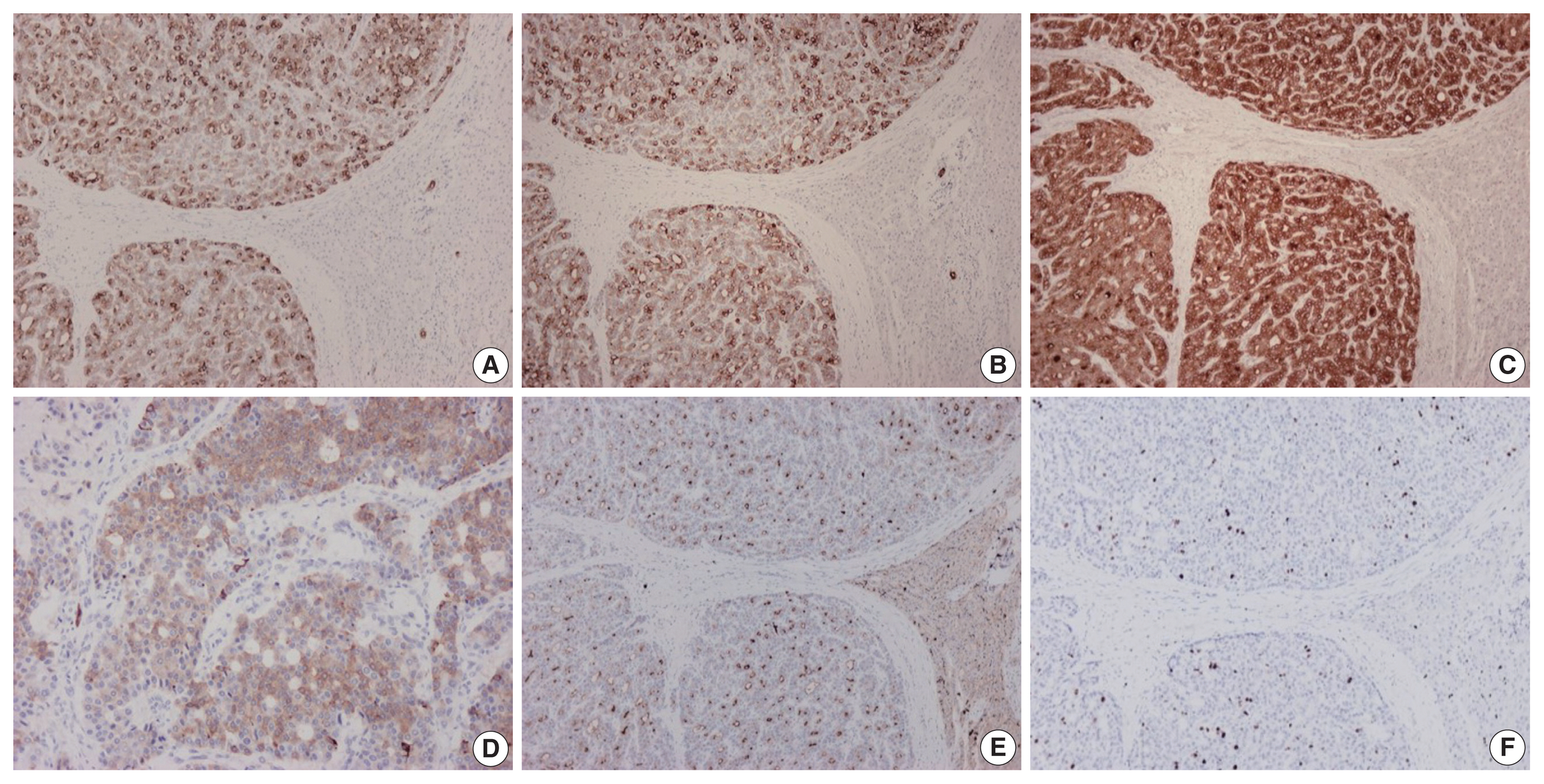
Immunohistochemical findings of tumor cells. The neoplastic cells are positive for cytokeratin 7 (A), cytokeratin 19 (B), and inhibin (C). Patchy mild positivity of tumor cells for synaptophysin is also seen (D). Stain for polyclonal carcinoembryonic antigen demonstrates membranous positivity around pseudoglandular structures (E). Ten percent of tumor cells are positive for Ki67 antigen (F). Adjacent hepatic parenchyma is seen on the right side in all photomicrographs except for D.
Further immunohistochemical stains for CD34 and α-caldesmon were performed to explore the vascular architecture of the tumor. A rich, thin-walled vascular bed was evident on CD34 stains (Fig. 3A), while sparse larger vessels with venous features were seen on stains for α-caldesmon (Fig. 3B).
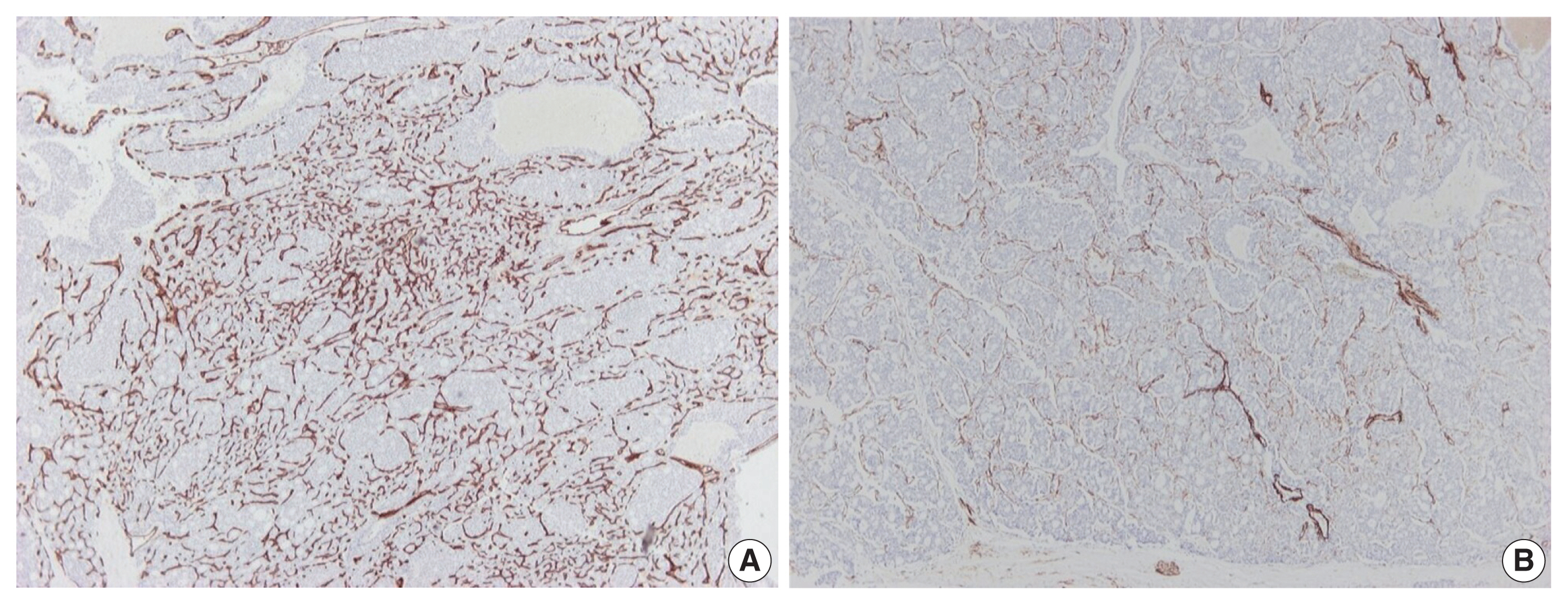
Immunohistochemical findings of tumor vessels. A rich thin-walled vascular bed is observed on CD34 stain (A), while stain for α-caldesmon shows sparse larger vessels with venous features (B).
DISCUSSION
HCEI is an enigmatic neoplasm that was originally recognized because of the combination of its unusual histologic features with the immunohistochemical expression of inhibin [1]. Sixteen cases of HCEI have been thus far reported, including the present one (Table 1) [1–6]. Twelve cases occurred in women and four in men; the age range of the patients was 17 to 54 years. HCEI has been found to exhibit varied architecture, with trabecular, pseudoglandular, follicular/microcystic, organoid, solid and tubular patterns of growth. The neoplastic cells diffusely express inhibin. Prognosis is variable. The patient of our original report [1] received no further treatment following surgical resection, and is well, without evidence of disease, 20 years after presentation. The patient of our current report is also alive and without evidence of disease 9 months after presentation. However, three of the other 14 patients reported to date have died of the disease, 5 had disease progression, one had stable disease, 3 were disease-free at last follow-up, and two were lost to follow-up (Table 1).

Reported cases of hepatic carcinoma expressing inhibin
Despite the small number of reported cases, there is sufficient evidence that HCEI is a distinct clinicopathologic entity [1–6]: (1) These tumors are relatively large (size range, 6.9 to 36 cm) when they become symptomatic, suggesting an indolent initial growth phase. (2) These tumors have characteristic histologic features, including trabecular, pseudoglandular, follicular/microcystic, organoid, solid and tubular patterns of growth. 3) In addition to inhibin positivity, HCEI characteristically shows positivity for ‘biliary’ markers (CK7 and CK19), mild or patchy positivity for neuroendocrine markers (synaptophysin, chromogranin, CD56), and absence of staining for hepatocellular markers (HepPar-1, arginase-1, glypican-3, α-fetoprotein). Albumin mRNA has also been detected in 10 cases by in situ hybridization [3–5]. (4) Molecular studies recently performed in three cases [5] have detected a novel NIPBL-NACC1 gene fusion in HCEI.
The histogenesis of HCEI remains elusive. Inhibin expression characterizes a limited range of normal cell types, including granulosa cells, luteinized thecal cells and hilus cells of the ovary, syncytiotrophoblastic cells and adrenocortical cells [1]. Theoretically, any of these cell types could be present in a heterotopic location, such as the liver, and give rise to tumors. However, tumors with the features of HCEI have not been described in the ovaries or adrenals. On the other hand, inhibin expression has occasionally been reported in tumor types without obvious connection to the normal cells mentioned above, such as mucinous cystic neoplasm of the liver and pancreas, as well as neuroendocrine tumors of clear cell type occurring in patients with von Hippel-Lindau disease [7]. It might be worth exploring whether there is any connection between these neoplasms and HCEI. Nevertheless, the possibility that inhibin expression may be triggered in cells with a biliary phenotype by NIPBL-NACC1 gene fusion cannot be ruled out.
From a practical point of view, it is possible that HCEI has been underreported, due to its histologic and immunohistochemical similarities with hepatocellular carcinoma, cholangiocarcinoma, primary or metastatic neuroendocrine tumors and metastatic follicular carcinoma of the thyroid gland. The diagnosis of HCEI can be easily missed if pathologists do not think of performing inhibin immunostain. Reports of additional cases, including further immunohistochemical and molecular studies, will be useful to define this proposed new entity with certainty. In this context, it would be of interest to assess inhibin expression in neoplasms that have histologic similarities with HCEI, such as primary hepatic neuroendocrine tumors (including a recently described “primary hepatic neuroendocrine tumor with unusual thyroid follicular-like morphologic characteristics”, which occurred in a 57-year old woman [8]), as well as tumors recently described as “thyroid-like intrahepatic cholangiocarcinoma”, which occurred in three women and one man, between the ages of 26 and 59 years [9–12].
In conclusion, HCEI is an emerging entity that pathologists should be aware of. This neoplasm can potentially cause diagnostic pitfalls by simulating histologically other primary or metastatic hepatic tumors. Performing inhibin immunostain could be of great help in the differential diagnosis of liver tumors with unusual histologic features.
Notes
Ethics Statement
Informed consent was obtained from the individual participant included in this study. In our institution, case studies are exempted from Institutional Review Board submission.
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: AS, EA, PH. Data curation: AS, EA, PH. Formal analysis: AS, EA, PH. Methodology: AS, EA, PH. Supervision: PH. Writing—original draft: AS, EA, PH. Writing—review & editing: AS, EA, PH. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.