Perspectives on single-nucleus RNA sequencing in different cell types and tissues
Article information

Abstract
Single-cell RNA sequencing has become a powerful and essential tool for delineating cellular diversity in normal tissues and alterations in disease states. For certain cell types and conditions, there are difficulties in isolating intact cells for transcriptome profiling due to their fragility, large size, tight interconnections, and other factors. Single-nucleus RNA sequencing (snRNA-seq) is an alternative or complementary approach for cells that are difficult to isolate. In this review, we will provide an overview of the experimental and analysis steps of snRNA-seq to understand the methods and characteristics of general and tissue-specific snRNA-seq data. Knowing the advantages and limitations of snRNA-seq will increase its use and improve the biological interpretation of the data generated using this technique.
The introduction of single-cell RNA sequencing (scRNA-seq) opened a new era in cell biology, where cellular identity and heterogeneity can be defined by transcriptome data [1]. One cell type with tremendous phenotypic and functional heterogeneity is neurons in the brain. Indeed, scRNA-seq has revealed diverse neuronal cell types in the mouse brain [2]. However, cell isolation by enzymatic tissue dissociation may damage neurons, and most human brain samples are not available as fresh tissues. Among the alternative approaches attempted, the isolation of single nuclei and subsequent RNA sequencing have enabled high-throughput transcriptome profiling at a single-cell resolution [3,4]. In addition to neurons, single-nucleus RNA sequencing (snRNA-seq) has been applied to diverse hard-to-dissociate tissues and cell types, including the kidney, heart, liver, adipocytes, and myofibers [5–9]. For most tissues, snRNA-seq is more powerful at recovering attached cell types, whereas scRNA-seq is biased towards immune cell types [5,10–12]. Moreover, the enzymatic dissociation required for scRNA-seq induces a stress response that alters the cellular transcriptome [9,10,13]. Using snRNA-seq can reduce cellular and stress response biases. The different gene expression fractions in the nucleus and cytoplasm make it necessary to generate snRNA-based data references, and these have recently been provided [14]. Combining scRNA-seq and snRNA-seq data will enable more comprehensive transcriptome profiling and cell-type annotation in tissues.
EXPERIMENTAL PROCEDURES FOR SINGLE-NUCLEUS RNA SEQUENCING
snRNA-seq was developed as a method to obtain transcriptome data from cells that cannot be successfully dissociated due to their size and/or fragility, such as neurons, adipocytes, and epithelial cell types from the kidney (Fig. 1A). Multi-nucleated cells, such as trophoblasts, osteoclasts, and skeletal myocytes, can be inimitably interrogated using snRNA-seq. Archived frozen tissues with broken cell membranes are also the primary targets of this method. The isolation of single nuclei, instead of whole cells, is achieved by cell membrane lysis, and nuclear transcriptome data are generated using scRNA-seq workflows (Fig. 1B). Both chemical and mechanical forces are used for cell membrane lysis. Competent buffers with nonionic detergents that disrupt cell membranes, but preserve nuclear membranes, have been tested for nuclear isolation [12]. Mechanical force is exerted using a Dounce homogenizer or other types of tissue lysers. To obtain high-quality nuclei containing transcripts, the buffers and wash conditions are important and need to be optimized for different tissue types. The inclusion of bovine serum albumin and a high concentration of RNase inhibitors, during and after the isolation process, is critical. After isolation, nuclear morphology indicative of intact nuclei are confirmed by microscopy at 40–60 × magnification (Fig. 1C). Overlysis results in clumping and poor transcript recovery, whereas under-lysis causes contamination by cytoplasmic RNAs.
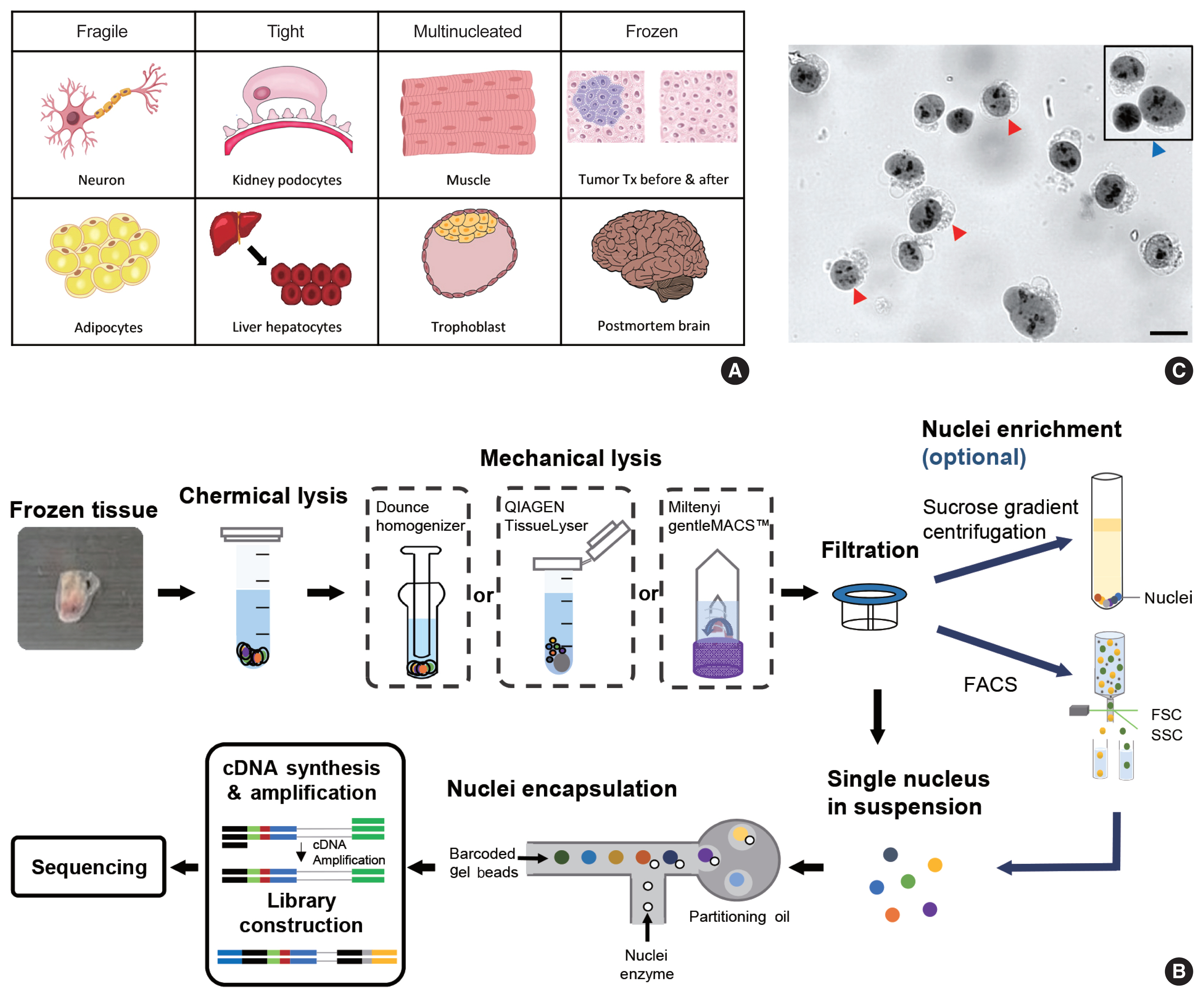
Summary of the single-nucleus RNA sequencing (snRNA-seq) experimental process. (A) Representative cell types and tissues fit for snRNA-seq–based transcriptome profiling. (B) Experimental workflow to isolate intact nuclei for snRNA-seq. Frozen tissue is dissected, chemically and mechanically lysed, and then filtered to obtain a single-nucleus suspension. Sucrose gradient centrifugation or flow cytometry analysis is used for nuclei enrichment (Optional). After reverse transcription and amplification, a cDNA library is constructed for sequencing. (C) Representative image of extracted nuclei stained with Trypan blue. High-quality (blue arrowhead) and poor-quality (red arrowhead) nuclei are marked. Scale bar = 20 μm. FACS, fluorescence-activated cell sorting; FSC, forward scatter; SSC, side scatter.
In addition to the isolation of intact nuclei, brain tissues require an additional clean-up process to remove excessive myelin debris. Iodixanol (OptoPrep, San Diego, CA, USA) or a sucrose gradient [15], a myelin removal column (Miltenyi, Bergisch Gladbach, Germany), and sorting by flow cytometry have been used for this extra clean-up process (Fig. 1B). The most frequently used buffer recipe for neurons in the brain is a combination of 250–320 mM sucrose and a low-concentration of non-ionic detergent, whereas the commercial EZ Prep Kit (Sigma, St. Louis, MO, USA) is the method of choice for kidney preparations. Sorting by flow cytometry is not recommended for kidney tissue. The use of commercial buffers other than EZ Prep, such as those from 10 × Genomics (Pleasanton, CA, USA) is also increasing, because of the minimal optimization requirement. Studies providing nuclear isolation protocols for snRNA-seq are listed in Table 1. The details of the buffer recipes and complete protocols can be found in these publications.
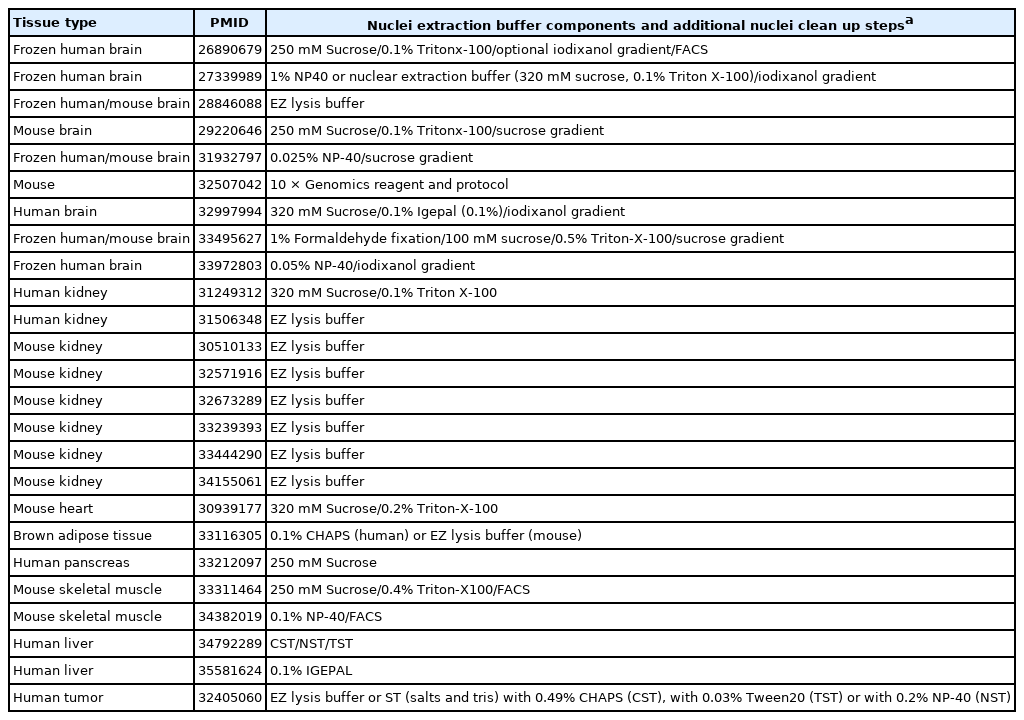
Representative studies reporting nuclei isolation protocols for the single-nucleus RNA sequencing
SINGLE-NUCLEUS RNA SEQUENCING DATA PROCESSING AND ANALYSIS
The data analysis pipeline for snRNA-seq is similar to the pipeline used for scRNA-seq (Fig. 2A). The most frequently used sequencing procedure for snRNA-seq is Chromium 3′ scRNA-seq (10 × Genomics), and the sequencing read mapping process (Cell Ranger 7.0, 10 × Genomics) currently used is identical for scRNA-seq and snRNA-seq. During this process, both exonic and intronic reads that map the sense orientation to a single gene are used for gene counting using the default option. In previous Cell Ranger versions, intronic mapped reads were not used for the default read count option in the scRNA-seq pipeline, and the option parameter, “--include-introns = true” needed to be added for snRNA-seq read counting. The inclusion of intronic reads in snRNA-seq is critical, as more than 50% of nuclear RNAs are typically intronic compared to 15%–25% of total RNAs [13,16]. Immune cell populations such as neutrophils and other granulocytes are more likely to be identified when intronic reads are included. Detection of neutrophils is difficult because of their low RNA content and low gene count [17]. Since neutrophils have a higher amount of introns compared to other cell types [18,19], the inclusion of intronic reads may enhance the recovery of neutrophils. According to the guideline by 10 × genomics, experimental steps are also important to enhance the neutrophil recovery such as immediate processing, sample preparation at room temperature, increasing polymerase chain reaction cycles during cDNA amplification, adding RNase inhibitors in the wash and suspension buffers, and enrichment by fluorescence-activated cell sorting into 0.04% bovine serum albumin solution in scRNA-seq [20].
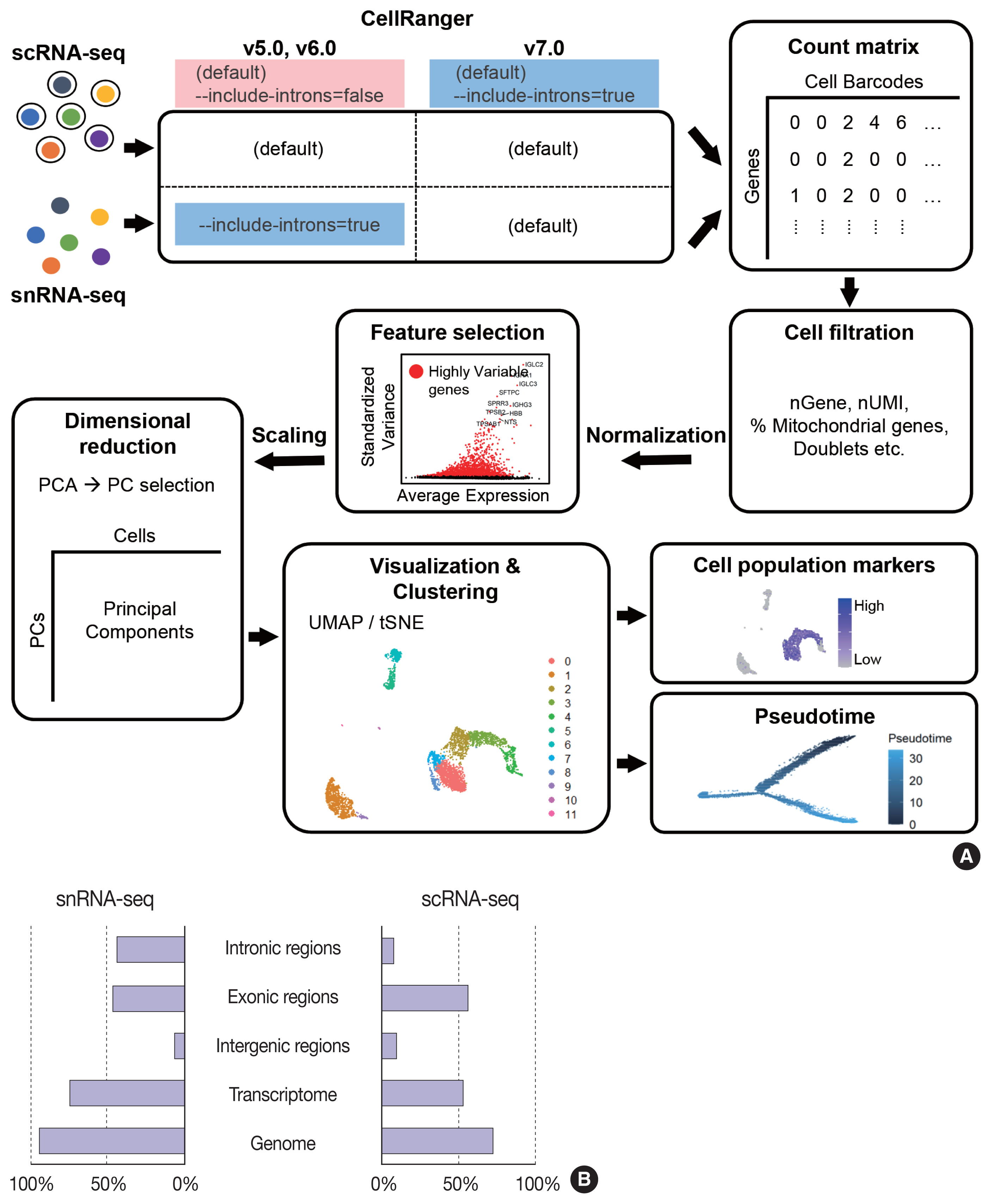
Summary of single-nucleus RNA sequencing (snRNA-seq) and single-cell RNA sequencing (scRNA-seq) analyses. (A) Schematic workflow of snRNA-seq and scRNA-seq analysis processes. (B) Distribution of confidently mapped snRNA-seq and scRNA-seq reads. Transcriptome, the fraction of reads mapped to the exons of an annotated transcript. Genome, fraction of reads mapped to exonic and non-exonic loci. PC, principal cells; PCA, principal component analysis; UMAP, uniform manifold approximation and projection.
From the filtered cell by gene matrices of snRNA-seq data, further quality control (QC) filtering, normalization, feature selection, scaling, dimensional reduction, and clustering can be performed for cell-type annotation, as in scRNA-seq data analyses. Mitochondrial or ribosomal gene contents, which are often used as QC parameters for scRNA-seq, are not robustly used in snRNA-seq, as mitochondria and ribosomes are excluded during the experimental procedure. The differences in sequencing reads between scRNA-seq and snRNA-seq are shown in Fig. 2B.
Differential expression analysis using bulk RNA sequencing data has demonstrated a high correlation between nuclei and whole-cell samples [21,22]. However, at the single-cell or single-nucleus levels, cell-to-cell or nucleus-to-nucleus correlations decrease and replicate variations become larger than the bulk samples [22]. Direct comparisons of matched scRNA-seq and snRNA-seq data from S1 cortex neurons have demonstrated differences in genomic read mapping to coding sequences, introns, or untranslated regions [23]. Significant gene length bias exists, such that nuclear-biased genes show a length of 17 kb compared with 188 kb for genes detected in both whole cells and nuclei. The total gene expression correlation between single-cell and single-nucleus data ranges from 0.21 to 0.74. In a study of adipocytes, the average gene expression correlation between whole-cell and nuclei data for white cells was found to be 0.5 or 0.6 (after normalization) [24]. Despite the relatively low correlations, diverse batch correction algorithms allow the co-clustering of identical cell types at a global scale in scRNA-seq and snRNA-seq data [24].
While data integration allows the combined clustering analysis of scRNA-seq and snRNA-seq data, direct comparisons of the two are difficult because of the differences in cellular and nuclear gene expression patterns. In addition, over-representation of immune cells by scRNA-seq and the superior representation of epithelial cell types by snRNA-seq suggest that complementary analysis is more appropriate than integrated analysis (Fig. 3).
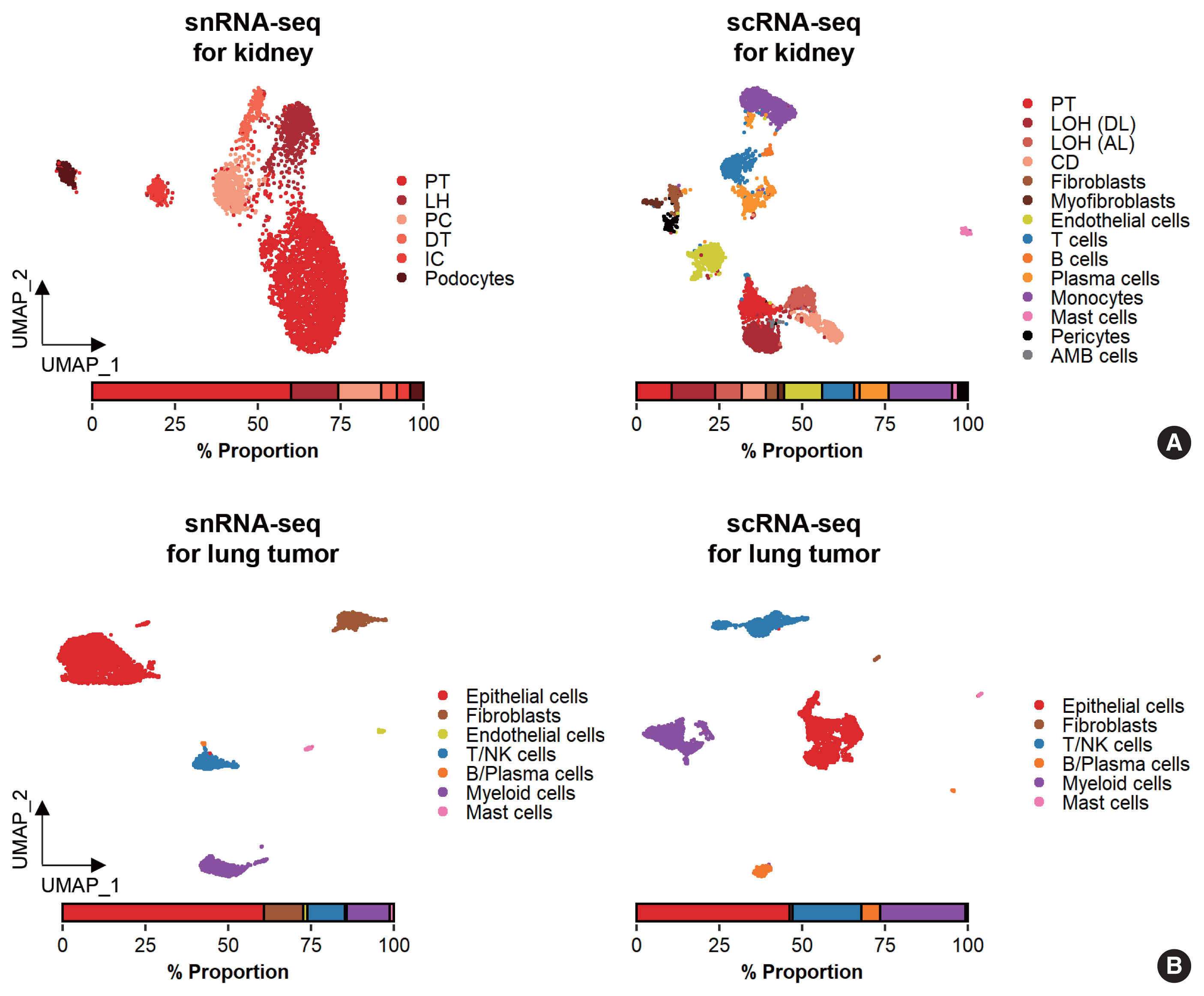
Comparison of cell types detected by single-nucleus RNA sequencing (snRNA-seq) and single-cell RNA sequencing (scRNA-seq). (A) Uniform manifold approximation and projection (UMAP) plots of snRNA-seq and scRNA-seq data for the human kidney. A bar plot representing the percentages of annotated nuclei and cell identities. AMB, ambiguous; CD, collecting duct; DT, distal tubule; IC, intercalated cells; LH, loop of Henle; LOH (AL), loop of Henle, ascending limb; LOH (DL), loop of Henle, distal limb; NK, natural killer; PC, principal cells; PT, proximal tubule. (B) UMAP plots of snRNA-seq and scRNA-seq data for lung tumors from a lung cancer patient.
SPECIFIC TISSUE OR CELL TYPE APPLICATIONS
Neurons in the brain
A high-throughput snRNA-seq protocol has been described for transcriptomic analysis of individual neurons from archived postmortem human brain tissues [25]. Before the introduction of high-throughput applications, low-throughput methods, such as intracellular tagging by transcription in vivo analysis [26] and extraction of the cytoplasmic contents using a glass microcapillary [27,28] or laser-capture microdissection [29] were explored, along with low-throughput snRNA-seq [22]. Lake and colleagues [4] applied snRNA-seq and identified 16 neuronal subtypes of the cerebral cortex from a postmortem brain.
Currently, snRNA-seq is extensively used to determine brain cell type complexity. The U.S. government’s Brain Research Through Advancing Innovative Neurotechnologies (BRAIN) Initiative [30] launched a project known as the BRAIN Initiative Cell Census Consortium to pursue a comprehensive human brain cell atlas [31]. These resources will serve as a reference for delineating brain functions and alterations in neurodegenerative and neurological diseases. To construct the brain cell atlas, electrophysiological, morphological, and transcriptional features were used for neuronal cell type specifications, signifying the importance of transcriptome-based cell type annotation in functional and anatomical contexts. Transcriptome-based neuronal identification was accomplished using both scRNA-seq and snRNA-seq [31] after regional dissection. Due to the under-representation of neuronal cell types in scRNA-seq data and the availability of frozen postmortem brains, more recent cell applications have concentrated on snRNA-seq.
Nonetheless, differences in the nuclear and cytoplasmic gene expression patterns, and limitations of snRNA-seq in the capture and characterization of non-neuronal cell types [32] necessitate the complementary use of scRNA-seq and snRNA-seq for cell type identification in the brain.
Epithelial cells in the kidney
Whereas the studies agree that average nephron number is approximately 900,000 to 1 million per kidney, numbers for individual kidneys range from approximately 200,000 to > 2.5 million [33]. Each nephron contains a glomerulus, which is a bundle of vessels through which waste materials are filtered from the blood. The glomerulus is enclosed in Bowman’s capsule, and filtered water, ions, and small molecules are collected in Bowman’s space. Podocytes in the epithelial lining of the Bowman’s capsule wrap around the capillaries of the glomerulus and leave filtration slits between them. Filtered materials leave Bowman’s space through a proximal tubule where reabsorption occurs. Epithelial cells lining the proximal tubule are covered with dense microvilli to facilitate transport. The modular characteristics of the kidney make biopsy an accessible and efficient sampling method for the characterization of the glomerulus.
Glomerular cell types in the kidney have been characterized using both scRNA-seq and snRNA-seq protocols. For the mouse kidney, an snRNA-seq experimental protocol yielded 20-fold more podocytes than an scRNA-seq protocol [9,34]. The Kidney Precision Medicine Project developed a reference tissue atlas for the human kidney with single-cell resolution and spatial context [35]. Rare epithelial cell types and states can be captured by snRNA-seq; however, immune components in the kidney are not well captured by snRNA-seq (Fig. 3A) [36]. Thus, the kidney atlas data incorporate snRNA-seq and scRNA-seq data for tissue atlas generation [37].
Tumors from frozen tissues
A diverse range of solid tumor tissues have been subjected to scRNA-seq, and the biological features of tumor cells and their surrounding microenvironments have been extensively studied. However, scRNA-seq data shows a heavy bias towards immune cell types when compared with bulk tissue data after cell type deconvolution (Fig. 3B). The use of snRNA-seq data may resolve this problem [12]. Side-by-side comparisons of scRNA-seq and snRNA-seq analyses of hepatocellular carcinoma [38] demonstrated the predominant capture of hepatocytes and carcinoma cells in snRNA-seq data compared with the immune cell-dominant landscape in scRNA-seq data. In a pancreatic cancer study, a combination of snRNA-seq and digital spatial profiling revealed that gene expression programs in malignant tumor cells and fibroblasts were enriched after chemotherapy and radiotherapy [39]. In addition to tumor-centric data analysis, snRNA-seq can be performed on longitudinal samples stored as frozen tissues. Similar to brain and kidney examples, immune cells in the tumor microenvironment can be efficiently captured by scRNA-seq.
CONCLUSION
Transcriptome-based cell type profiling by scRNA-seq has remarkably enhanced our understanding of cellular diversity. While scRNA-seq shows good performance at capturing immune cell diversity, the cellular landscape depicted is biased against for attached cell types and is missing fragile cells. In most tissues, snRNA-seq can be used to obtain more information about these cell types, including epithelial cells, fibroblasts, neurons, and adipocytes. In addition, snRNA-seq can be used for frozen tissues, such as postmortem brain and archived tumor samples. After the successful isolation of nuclei, experimental and analysis pipelines used for scRNA-seq can be adopted for snRNA-seq. In the analysis, data from the two methods should be combined with caution, considering the differences in cellular and nuclear RNA gene expression patterns.
Notes
Ethics Statement
Not applicable.
Availability of Data and Material
The datasets analyzed during the current study are available in the Gene Expression Omnibus repository at GSE137444 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE137444], GSE114156 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114156], and GSE109564 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109564].
Code Availability
Not applicable.
Author Contributions
Conceptualization: SAY, HOL. Data curation: NK. Funding acquisition: HOL. Investigation: NK, HK, AJ. Supervision: SAY, HOL. Visualization: NK, HK, AJ. Writing—original draft: NK, HK, SAY, HOL, AJ. Writing—review & editing: NK, HK, SAY, HOL, AJ. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
This study was supported by the Bio & Medical Technology Development Program of the National Research Foundation, funded by the Ministry of Science and ICT (NRF-2019M3A9B6064691).