Clinical practice recommendations for the use of next-generation sequencing in patients with solid cancer: a joint report from KSMO and KSP
Article information
 , Hyo Sup Shim2, Sheehyun Kim3, In Hee Lee4, Jihun Kim5, Shinkyo Yoon6, Hyung-Don Kim6, Inkeun Park6, Jae Ho Jeong6, Changhoon Yoo6, Jaekyung Cheon6, In-Ho Kim7, Jieun Lee7, Sook Hee Hong7, Sehhoon Park8, Hyun Ae Jung8, Jin Won Kim9, Han Jo Kim10, Yongjun Cha11, Sun Min Lim12, Han Sang Kim12, Choong-Kun Lee12, Jee Hung Kim13, Sang Hoon Chun14, Jina Yun15, So Yeon Park16, Hye Seung Lee17, Yong Mee Cho5, Soo Jeong Nam5, Kiyong Na18, Sun Och Yoon2, Ahwon Lee19, Kee-Taek Jang20, Hongseok Yun3, Sungyoung Lee3, Jee Hyun Kim9,, Wan-Seop Kim21,
, Hyo Sup Shim2, Sheehyun Kim3, In Hee Lee4, Jihun Kim5, Shinkyo Yoon6, Hyung-Don Kim6, Inkeun Park6, Jae Ho Jeong6, Changhoon Yoo6, Jaekyung Cheon6, In-Ho Kim7, Jieun Lee7, Sook Hee Hong7, Sehhoon Park8, Hyun Ae Jung8, Jin Won Kim9, Han Jo Kim10, Yongjun Cha11, Sun Min Lim12, Han Sang Kim12, Choong-Kun Lee12, Jee Hung Kim13, Sang Hoon Chun14, Jina Yun15, So Yeon Park16, Hye Seung Lee17, Yong Mee Cho5, Soo Jeong Nam5, Kiyong Na18, Sun Och Yoon2, Ahwon Lee19, Kee-Taek Jang20, Hongseok Yun3, Sungyoung Lee3, Jee Hyun Kim9,, Wan-Seop Kim21,
Abstract
In recent years, next-generation sequencing (NGS)–based genetic testing has become crucial in cancer care. While its primary objective is to identify actionable genetic alterations to guide treatment decisions, its scope has broadened to encompass aiding in pathological diagnosis and exploring resistance mechanisms. With the ongoing expansion in NGS application and reliance, a compelling necessity arises for expert consensus on its application in solid cancers. To address this demand, the forthcoming recommendations not only provide pragmatic guidance for the clinical use of NGS but also systematically classify actionable genes based on specific cancer types. Additionally, these recommendations will incorporate expert perspectives on crucial biomarkers, ensuring informed decisions regarding circulating tumor DNA panel testing.
Over the past few years, next-generation sequencing (NGS)–based genetic testing has emerged as a crucial aspect of cancer patient care, with the number of tests performed rapidly increasing since its reimbursement by the national health insurance in Korea in 2017. However, as the use of NGS-based genetic testing continues to expand, there is an increasing need for maximizing benefits for patients while also considering cost-effectiveness.
The primary objective of NGS-based genetic testing is to identify targetable actionable genes that can guide treatment selection. However, its application has expanded to include diagnosis and exploration of resistance mechanisms, enabling more personalized treatment options. Moreover, biomarkers like homologous recombination deficiency (HRD), microsatellite instability–high (MSI-H)/mismatch repair deficiency (MMR-D), and high tumor mutational burden (TMB-H) have gained increasing significance. Consequently, NGS-based testing is now widely used to analyze these biomarkers and make well-informed treatment decisions.
With the expanding application of NGS-based genetic testing, there is a need for expert consensus on best practices and guidelines for its use. This recommendation aims to (1) provide guidance on the practical application of NGS in daily clinical practice and (2) classify actionable gene lists by cancer type, based on a comprehensive review of the literature and the consensus of experts. Furthermore, the recommendation will present expert opinions, based on existing evidence, regarding biomarkers including HRD, MSI-H/MMR-D, TMB, and circulating tumor DNA (ctDNA) panel testing.
MATERIALS AND METHODS
The Korean Society of Medical Oncology (KSMO) and the Korean Society of Pathologists (KSP) have collaborated to develop subsequent clinical practice recommendations. These focus on key questions not addressed in the previous guidelines for NGS-based genetic testing and the molecular tumor board from the KSMO and Korean Cancer Study Group (KCSG) Precision Medicine Networking Group [1]. In March and April of 2022, the Steering Committee and Writing Committee were reestablished. They were comprised of medical oncologists, pathologists, and bioinformaticians convened by KSMO, KCSG, and KSP. Two main issues were addressed: the proper recommendations for NGS-based genetic testing in solid cancers, and the classification level determination of genes applicable in Korea. The committees initially conducted a survey to assess the appropriateness of key questions, achieving consensus through feedback from all committee members, to confirm the final selection of key questions. Subsequently, recommendations for these questions were drafted by the Steering Committee and further refined through extensive discussions with all committee members during a comprehensive workshop in September 2022. These modified recommendations were then finalized through a final survey in November 2022. Additionally, the Writing Committee classified actionable genes by cancer type using the Korean Precision Medicine Networking Group (KPMNG) scale for clinical actionability of molecular targets (Table 1). The references for determining the actionability of target genes include case series and clinical trials from all phases (phase I, II, III) published up to August 31, 2023. Studies that were part of basket trials were also considered for inclusion. Furthermore, significant abstracts from clinical trials presented at the American Society of Clinical Oncology Annual Meeting and the European Society for Medical Oncology (ESMO) Congress were incorporated. Subsequently, these gene lists, along with their corresponding references, were shared with disease-specific divisions within KCSG and KSP, where feedback and input from these committees were incorporated to further refine the rankings. The lists underwent one final review and confirmation by the entire committee. The finalized recommendations were presented at the 2023 KSMO annual meeting and announced at the 2023 KSP annual meeting. These recommendations have received endorsements from both KSMO and KSP.

KEY QUESTIONS AND RECOMMENDATIONS
Question 1. What are the appropriate recommendations for NGS-based genetic testing in solid cancers?
Recommendation 1. NGS-based genetic testing is recommended for patients with advanced or metastatic solid cancers who are eligible for systemic treatments.
There is mounting evidence that NGS-based matched treatments enhance outcomes in patients with advanced or metastatic cancers [2-6]. Even in tumor types like breast cancer, where the role of NGS has traditionally been less defined, a recent study has shown improved treatment outcomes when patients were matched to appropriate therapies through comprehensive genomic analysis, including NGS [7].
Genomic testing should be conducted in patients with advanced or metastatic solid cancers if there are approved treatments matching genomic biomarkers by a regulatory authority. For instance, several genetic tests, including those for EGFR, ALK, ROS1, BRAF, MET, KRAS, ERBB2, and RET, should be conducted in patients with non-squamous non–small cell lung cancer (NSCLC). In cases where multiple gene tests are required, NGS can efficiently utilize tumor tissue compared to testing individual genes. The National Comprehensive Cancer Network guideline for NSCLC also recommends panel-based genomic testing by NGS [8]. The use of a multi-gene panel by NGS is also recommended for tumors like ovarian cancer, prostate cancer, and pancreatic cancer. Testing for homologous recombination repair (HRR) related genes is required for these types of cancers to inform the use of poly(ADP-ribose) polymerase (PARP) inhibitors. Even for patients with cancers in which actionable genetic alterations are rarely found, NGS is recommended, taking into account tumor-agnostic biomarkers. MSI-H/MMR-D, TMB-H, BRAF V600E, RET fusion, and NTRK fusions have been approved by the U.S. Food and Drug Administration (FDA) as tumor-agnostic biomarkers [9-20]. In Korea, matched treatments for tumors with MSI-H/MMR-D and NTRK fusions have been approved.
If a biomarker-matched treatment showing clinical benefit has not yet received regulatory approval, we strongly encourage patients to participate in clinical trials based on molecular profiles from NGS. Our goal is to provide maximum treatment options for individual patients with advanced or metastatic cancer. The probability of detecting actionable genetic alterations using NGS varies based on the cancer type [2]. Given that the potential benefits of NGS may vary among individuals, it is essential to discuss its aims and limitations with the patient. Furthermore, NGS is not recommended when systemic treatment is unfeasible due to factors including the patient’s performance status, comorbidities, and socioeconomic conditions.
Recommendation 2. NGS-based genetic testing can be recommended for the pathological diagnosis of solid cancers.
Precise pathological diagnosis is a fundamental component of precision oncology and in predicting prognosis for patients with solid cancer. Notably, in the recently published classification of tumors by the World Health Organization (WHO), the diagnosis of tumors defined by genetic alterations is gradually expanding. Consequently, there are increasing cases in which a final pathological diagnosis is made based on NGS results. In addition, OncoKB [21], which is widely referred to in the interpretation of genetic alterations, provides information about diagnosis of hematologic malignancy by classifying the genetic alterations into ‘Diagnostic’ Level Dx1 (required for diagnosis), Dx2 (supports diagnosis), and Dx3 (investigational diagnosis). It is anticipated that this trend will soon be reflected in the diagnosis of solid cancers. We will briefly discuss the application of NGS in the diagnosis of bone and soft tissue sarcoma, renal cell carcinoma, and central nervous system tumors, using these as representatives.
Bone and soft tissue sarcomas
As more than half of soft tissue tumors and approximately a quarter of bone tumors harbor recurrent genetic alterations [22], molecular analysis is a strong diagnostic tool for the evaluation of bone and soft tissue sarcomas. There are several advantages of using NGS: simultaneous examination of multiple genomic regions, low-level tumor sample requirement and intuitive visualization of results [23]. NGS panels designed for sarcoma diagnosis utilize primers for the detection of fusions, amplifications, deletions and point mutations, which broadly cover genetic alterations in various sarcoma types. In daily practice, pathologists often encounter cases in which NGS provides the precise diagnosis by confirming or excluding differential diagnoses. Some cases can be even diagnosed toward unsuspected entities on the microscopic examination after NGS analysis [24].
NGS analysis may be applied for differential diagnosis of bone and soft tissue sarcomas as follows: (1) low-grade central osteosarcoma (MDM2) vs. fibrous dysplasia (GNAS); (2) chondroblastic osteosarcoma (chromosomal instability) vs. chondrosarcoma (IDH1/2); (3) malignant peripheral nerve sheath tumor (CDKN2A) vs. atypical neurofibroma; (4) liposarcoma (MDM2) vs. atypical pleomorphic lipomatous tumor (RB1); (5) alveolar rhabdomyosarcoma (PAX3/7::FOXO1) vs. embryonal rhabdomyosarcoma (mutations in RAS-MAPK pathway); (6) tumors of uncertain differentiation (Ewing sarcoma, round cell sarcoma with EWSR1-non-ETS fusions, CIC-rearranged sarcoma, sarcoma with BCOR genetic alterations, synovial sarcoma, alveolar soft part sarcoma, extraskeletal myxoid chondrosarcoma, clear cell sarcoma of soft tissue, etc.)
Renal cell carcinoma
NGS-based genetic panel test can be recommended for the pathological diagnosis of molecularly defined renal cell carcinoma (RCC), which includes fumarate hydratase (FH)–deficient RCC, succinate dehydrogenase (SDH)–deficient RCC, TFE3-rearranged RCC, TFEB-rearranged or TFEB-amplified RCC, ELOC (formerly TCEB1)-mutated RCC, SMARCB1 (INI1)- deficient RCC, and ALK-rearranged RCC according to the recent 2022 WHO classification [25]. The molecular alterations of these renal tumors are as follows: biallelic FH mutation/inactivation in FH-deficient RCC; inactivating mutations of one of SDH genes, most commonly SDHB, followed by SDHA and SDHC, and rarely SDHD in SDH-deficient RCC; translocations involving TFE3 in TFE3-rearranged RCC; translocations involving TFEB in TFEB-rearranged RCC; TFEB amplification in TFEB-amplified RCC; inactivating mutations exclusively at TCEB1 Y79 in ELOC (formerly TCEB1)-mutated RCC; translocations or deletions involving 22q11.23 in SMARCB1 (INI1)-deficient RCC; translocations involving ALK in ALK-rearranged RCC. In addition, NGS-based genetic panel test may also be recommended for morphologically defined renal tumors with characteristic molecular alteration. Clear cell RCC is characterized by the loss of chromosome 3p accompanied by the inactivation mutation or methylation of the remaining VHL gene. Papillary RCC commonly shows gains of chromosomes 7 and 17, and loss of the Y chromosome with MET alterations in the low-grade tumor. Chromophobe RCC has losses of multiple chromosomes including 1, 2, 6, 10, 13, 17, 21, and Y. Eosinophilic solid and cystic RCC can show TSC gene mutations or biallelic losses.
Central nervous system tumor
With the development of research techniques such as NGS, our understanding of the molecular and clinicopathological characteristics of brain tumors has advanced greatly. Based on these changes, following the 2016 Central Nervous System (CNS) WHO classification revised 4th edition [26] and cIMPACT-NOW [27], the 2021 CNS WHO classification 5th edition [28] fully included the molecular genetic characteristics of tumors in the WHO classification of brain tumors. In the 2021 CNS WHO classification, several molecular genetic characteristics such as gliomas, glioneuronal tumors, ependymomas, embryonic tumors (medulloblastoma, etc.), and meningiomas were introduced into the diagnostic criteria. Molecular genetic characteristics included in the diagnostic criteria range from those that can be identified with a single test (sequencing, fluorescence in situ hybridization, etc.) to those that require integrated identification of various genes involved in a specific pathway, as well as those that identify chromosomal arm-level copy number alterations. To cover all of these, NGS testing is essential. In addition, these molecular classifications determine the diagnosis of the tumor and further determine the WHO grade, which is a basic brain tumor grading system that determines the treatment strategy. The use of traditional histopathological morphological classification alone without NGS testing can mislead patients’ treatment strategies.
Recommendation 3. NGS-based genetic testing can be repeated in patients with solid cancer in case of disease recurrence or development of drug resistance.
Acquired resistance inevitably occurs with the growing use of targeted agents targeting various driver oncogenes. Representatively, we have seen the successful development of osimertinib, the third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) during the last decade [29]. At the time of drug development, osimertinib was developed for the patients who revealed the acquired EGFR threonine to methionine at codon 790 (T790M) mutation at the time of treatment failure with first- or second-generation EGFR TKI [30]. Therefore, the detection of EGFR T790M has been crucial for making treatment decisions in patients who experienced treatment failure with first- or second-generation EGFR TKIs [8]. Apart from EGFR T790M, other types of acquired resistance mechanisms were revealed by NGS, such as ERBB2 amplification or MET amplification [31]. Given the recent memorial imprint of resistance mechanism discovery, we have started using repeated NGS to detect acquired resistance in on-treatment tumor tissue, as well as in liquid biopsy samples.
Generally, acquired resistance can be classified into two categories: (1) target-dependent, such as target gene mutations, and (2) target-independent, such as gene aberrations in bypass pathways [32]. Beyond the EGFR T790M mutation, the EGFR C797S mutation is one of the most common EGFR-dependent resistance mechanisms against osimertinib [33]. MET amplification is another type of bypass pathway resistance mechanism across oncogene-driven subsets of NSCLC [34]. The EML4::ALK fusion, occurring in 3%–7% of all NSCLC cases, is currently treated with alectinib or brigatinib, the second-generation ALK TKIs, which are the standard treatments for treatment-naïve ALK-positive NSCLC patients [35-37]. ALK G1202R, solvent front mutation affecting drug binding to active site, is the most common target-dependent mutation [38]. Detecting the ALK G1202R mutation through NGS enables the prediction of a notable response with subsequent lorlatinib. NTRK fusion is a tumor agonistic driver oncogene, detected in less than 1% of solid cancers. With introduction of larotrectinib and entrectinib in clinic, several target-dependent point mutations were noted, which can be found by NGS [19,20]. Repotrectinib (TPX-0005) has demonstrated anti-tumor efficacy in patients previously treated with NTRK-targeting TKIs and who harbor target-dependent TRK mutations [39].
Since the 2000s, the clinical use of NGS has expanded beyond the detection of driver oncogenes. It has paved the way for the discovery of novel targets associated with acquired resistance and provided valuable insights into potential targets for the next generation of targeted therapeutics. However, it’s important to acknowledge certain limitations associated with the repetition of NGS testing. Challenges include the increased cost, difficulties in obtaining repeated tumor biopsies, and associated risks. Additionally, the likelihood of identifying actionable targets at the point of resistance can vary depending on the specific cancer type and drugs, with potential restrictions in drug availability. Nonetheless, it remains evident that NGS can play a crucial role in helping inform subsequent treatment decisions for certain patients who have experienced treatment failure with targeted therapy.
Question 2. How can we determine the classification level of genes applicable in Korea?
Advancements in NGS technologies have facilitated the identification of driver mutations in cancer, prompting a shift from a histology-based to a molecular-based approach in cancer treatment. Simultaneously, the advent of targeted therapies has allowed for treatments based on genetic alterations irrespective of the tumor’s origin. This concept, known as tissue-agnostic indication, has demonstrated promising results in recent studies and has become a crucial element in the standard care for cancer. Currently, the tissue-agnostic indications approved by the FDA are listed in Table 2 [9-20,40].
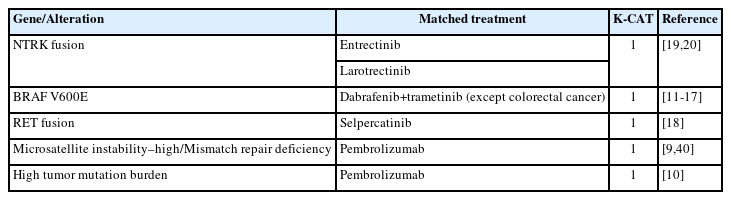
List of genetic alterations with tumor agnostic indications by FDA
Taking into account both the evidence level of clinical research and clinical benefit, the committee members classified actionable genes for each type of cancer based on their level using KPMNG scale of Clinical Actionability of molecular Targets (K-CAT). We also included certain genes, such as POLE in endometrial cancer, that are clinically significant and thus necessitate testing. The actionable gene lists for NSCLC, breast cancer, esophageal cancer, stomach cancer, colorectal cancer, head and neck cancer, pancreatic cancer, biliary tract cancer, endometrial cancer, urothelial cancer, and kidney cancer are provided in Tables 3–17 [11-15,29,36,37,41-190]. Each table included genes corresponding to levels 1 through 3A.

List of genomic alterations level 1/2/3A according to K-CAT in advanced NSCLC
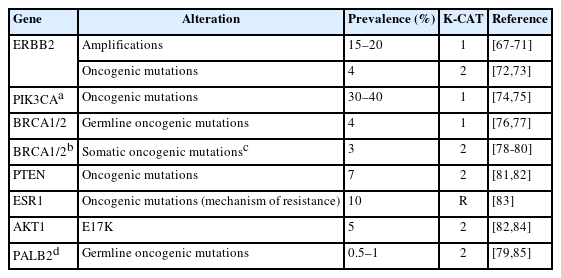
List of genomic alterations level 1/2/3A according to K-CAT in advanced breast cancer

List of genomic alterations level 1/2/3A according to K-CAT in advanced esophageal cancer
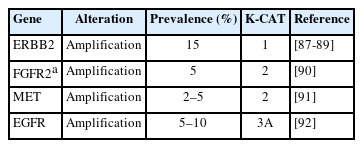
List of genomic alterations level 1/2/3A according to K-CAT in advanced stomach cancer

List of genomic alterations level 1/2/3A according to K-CAT in advanced colorectal cancer
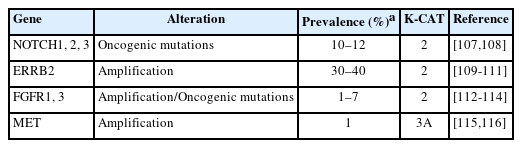
List of genomic alterations level 1/2/3A according to K-CAT in advanced head and neck cancer

List of genomic alterations level 1/2/3A according to K-CAT in advanced pancreatic cancer

List of genomic alterations level 1/2/3A according to K-CAT in advanced biliary tract cancer
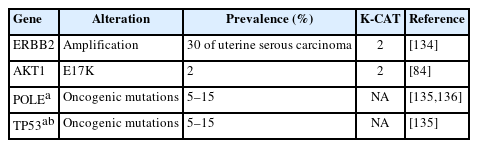
List of genomic alterations level 1/2/3A according to K-CAT in advanced endometrial cancer
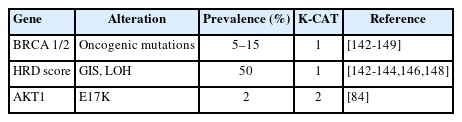
List of genomic alterations level 1/2/3A according to K-CAT in advanced ovarian cancer
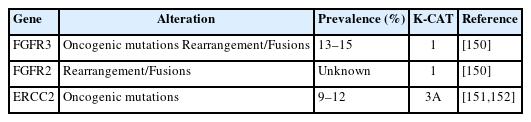
List of genomic alterations level 1/2/3A according to K-CAT in advanced urothelial cancer

List of genomic alterations level 1/2/3A according to K-CAT in advanced prostate cancer
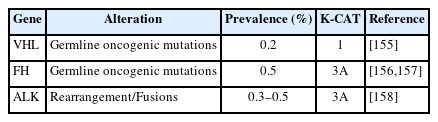
List of genomic alterations level 1/2/3A according to K-CAT in advanced kidney cancer
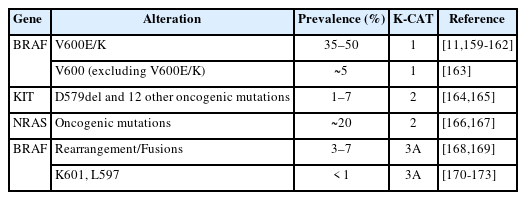
List of genomic alterations level 1/2/3A according to K-CAT in advanced melanoma

List of genomic alterations level 1/2/3A according to K-CAT in advanced sarcoma
Additional topics
Homologous recombination deficiency
Genomic instability is one of the most frequent underlying features of carcinogenesis, and defective DNA repair has been described as a cancer hallmark [191]. HRR is a series of interrelated pathways that function in the repair of DNA double-strand breaks and interstrand crosslinks [192]. Important genes involved in the HRR process include BRCA1, BRCA2, RAD51, RAD51C, RAD51D, ATM, ATR, PALB2, MRE11, NBS1, BARD1,CHEK1, and CHEK2 [193,194]. However, it is essential to note that the list of genes known to be related to the HRR process is continually evolving through ongoing research. A defect in the HRR pathway has been linked to several cancers, including breast, ovarian, prostate and pancreatic cancer [117,142,153,195], and HRD can make tumors more sensitive to platinum-based chemotherapy and PARP inhibitors [196,197]. Thus, it is critical to develop methods for determining the HRD status in order to maximize clinical benefit from these drugs.
There are three main categories of available tests for HRD analyzing (1) the etiology of HRD (mutation/methylation sequencing), (2) the current homologous recombination status (functional assays), and (3) prior HRD exposure (genomic scars). Each type of cancer (ovarian, breast, pancreatic and prostate) requires different tests. The germline BRCA 1/2 mutation test is useful for predicting response to PARP inhibitors in ovarian and breast cancer [76,143-146,198]. In ovarian cancer, tumor (incorporating germline and somatic) as well as somatic BRCA 1/2 mutation testing exhibit good clinical validity by reliably identifying the subset of patients who benefit from PARP inhibitor therapy [146-148]. Evidence regarding the benefit of mutation tests for each non-BRCA HRR gene for predicting responses to PARP inhibitors remains insufficient in ovarian cancer. HRD tests using genomic instability scores (GIS) or loss of heterozygosity (LOH) scores are useful for predicting the responses to PARP inhibitors in ovarian cancer patients without BRCA 1/2 mutation [142,144,146]. The GIS from myChoice CDx (Myriad Genetics) represents the sum of LOH, large-scale transitions, and telomeric allelic imbalance and a GIS of 42 has been established as the threshold to determine HRD positivity [199,200]. To date, GIS is the only genomic scar assay that has been evaluated in first-line randomized controlled trials for ovarian cancer [142,143]. The LOH test (FoundationOne CDx, Foundation Medicine) uses NGS to determine the percentage of genomic LOH and LOH-high is defined with a cut-off of 16% or higher, referencing The Cancer Genome Atlas data [201]. In metastatic pancreatic cancer, a germline BRCA 1/2 mutation test is recommended to evaluate the potential benefits of PARP inhibitors as maintenance treatment for patients whose tumors have not progressed after first-line platinum-based chemotherapy [117]. In castration-resistant prostate cancer, it is recommended to assess by sequencing for BRCA 1/2 mutations, at a minimum, using germline and/or somatic tumor DNA [153,202]. To date, insufficient evidence is available regarding the benefit of performing a HRD functional assays to predict response to PARP inhibitor; however, the potential for using functional assays in conjunction with HRR gene tests and genomic tests should be evaluated. While there have been multiple NGS assays to evaluate HRD status, only a limited number of tests are clinically accepted, and their technical details including evaluation criteria are unclear. Many methodological approaches have been proposed to measure HRD status using NGS data of various types, including whole genome sequencing (WGS), whole exome sequencing (WES) and targeted sequencing [203,204]. However, the absence of congruent measure remains a challenge to validate their reliability and consistency. Although WGS has not yet been approved for the diagnosis of HRD, it might become a promising diagnostic tool for HRD in the near future.
Microsatellite instability-high/mismatch repair deficiency
MSI-H/MMR-D has become an important biomarker of eligibility for immune checkpoint inhibitor (ICI) therapy as the FDA has approved ICIs for patients with unresectable or metastatic MSI-H/MMR-D solid cancers regardless of tumor types [9,40,205]. The polymerase chain reaction (PCR)–based assessment of selected microsatellite loci in a patient’s tumor and matched non-neoplastic tissue had been accepted as the gold standard method before the era of NGS. Nevertheless, the PCR-based MSI test can be misleading in certain cases because the selected microsatellite loci (typically, 5 to 8 loci) may not cover all affected microsatellite regions [206]. Alternatively, MMR-D can be inferred through immunohistochemistry (IHC) of MMR proteins, such as MLH1, MSH2, MSH6, and PMS2, since most MMR-deficient tumors exhibit a loss of MMR protein expression. However, there are limitations to detecting MMR-D by the IHC method. Certain tumors harboring pathogenic missense or in-frame insertion/deletion mutations of MMR genes may still show intact MMR protein expressions, and interpretation errors may occur when the staining quality is poor.
Since NGS is now widely used in clinical practice, it has been investigated whether NGS can be used to detect MSI-H/MMR-D in clinical setting. Numerous validation studies have demonstrated that NGS can accurately detect pathogenic or likely pathogenic mutations affecting MMR genes and can determine MMR-D reliably. Thus, there is a consensus that NGS can replace the standard PCR-based MSI test. NGS can detect MSI-H/MMR-D in various ways [207]. Several computational tools for detection of MSI-H/MMR-D using NGS data are available: mSINGS [208], MSIsensor [209], MANTIS [210], and MOSAIC [211]. Furthermore, NGS can detect MSI-H/MMR-D even in the absence of the patient’s matched normal tissue [212,213]. Furthermore, pathogenic or likely pathogenic MMR gene mutations detected by NGS testing may select candidates of germline genetic testing for Lynch syndrome. Finally, NGS-based MSI-H/MMR-D testing may provide information about eligibility for immunotherapy in tumor types where MMR IHC and/or PCR-based MSI tests have not been done during routine clinical practice.
Analysis of TMB by NGS panel
ICIs can enhance a durable anti-tumor immune response and prolong overall survival [214]. However, only a subset of the patients showed a dramatic response to immunotherapy, and the identification of predictive biomarkers was essential to identify responders to immunotherapy, such as programmed death-ligand 1 expression, MSI-H/MMR-D and TMB-H [215-217]. TMB is defined as the number of somatic mutations (mut) per megabase (Mb) of genomic sequence [217]. TMB is a surrogate marker for making immunogenic neopeptides shown on the surface of tumor cells by major histocompatibility complexes, which affect the anti-tumor immune response to ICIs [218,219].
In June 2020, the FDA authorized pembrolizumab for the treatment of adult and pediatric patients with unresectable or metastatic TMB-H (≥10 mut/Mb) solid tumors, as determined by FoundationOneCDx assay, that have progressed following prior treatment and who have no satisfactory alternative treatment options [220]. Therefore, determining the TMB value and identifying TMB-H tumors are among the most critical aspects in the clinical NGS analysis.
Although the TMB calculation can vary according to the test assays, the gold standard method for TMB estimation is WES with tumor tissues and matched normal samples. However, since WES has limitations in terms of time and costs to apply in clinical use, analytic methods and algorithms have been developed for calculating TMB from clinical targeted NGS panel tests [221,222]. Targeted NGS panel tests usually cover only a small limited size (about 1 to 2 Mb) of exonic regions, so sophisticated bioinformatic algorithms and statistical methods must be applied to filter out noise variants and artifacts caused by formalin-fixed tissues. For tumor-only sequencing, which is currently conducted in most targeted gene panels in Korea, germline variants are filtered out using genomic information from public databases or data on allele frequency in normal populations to avoid TMB overestimation. In several studies, the evaluated TMB from targeted NGS panel testing showed a high correlation with the TMB calculated by WES using analytic techniques [221,222].
Since the targeted gene panels currently used in the clinic have different analysis pipelines for variant calling and apply various filtering criteria to select variants used in TMB calculation, TMB values vary among the tests, and the criteria for TMB-H are diverse [223]. Also, the distribution of TMB values and criteria for TMB-H are different by tumor type, even when calculating TMB with the same panel. In general, more than TMB of 10 mut/Mb has been used for the definition of TMB-H tumors, but the reliable value of TMB-H can be different among the test panels and requires caution in interpreting the estimated TMB value. In some studies, the TMB of 17–20 mut/Mb is considered TMB-H and a candidate for immunotherapy conservatively [224]. Therefore, standardization of TMB analysis among test panels, validation of TMB-H tumors with different assays, and establishing reliable criteria for TMB-H will be needed for the further precise application of TMB analysis with the clinical tumor NGS panels.
Clinical utility and limitations of ctDNA-based genetic panel tests using blood sample
As the growing number of druggable oncogenic drivers has been identified in solid cancer [225], ctDNA-based approach can be used as an alternative approach for facilitating the identification of tumor tissue genotype. However, ctDNA can be influenced by multiple preanalytical factors and the methodology of analysis [226]. Since the ctDNA detection rate is highly related to tumor burden and is affected by various factors such as plasma levels of ctDNA, assay sensitivity, and tumor biology, a negative result from the ctDNA test may not necessarily indicate a true negative. In particular, low analytical sensitivity may occur because ctDNA assay are performed solely on DNA derived from tumor cells [227]. Recent studies have reported that gene fusions and splice variants have higher detection rates when sequencing is performed with RNA transcripts [228,229]. In addition, in the case of copy number variations (CNVs), determining the presence of CNVs remains challenging due to its dependence on ctDNA fractions [230,231]. Hence, ctDNA-based test reports should include essential elements, including pre-analytical elements, sequencing results, potential factors related to the germline variants, and limitations of assays to assist the interpretation of the report to the clinician [232].
ctDNA-based genotyping can be used as either complementary to tissue genotyping or as the first choice in certain circumstances. ctDNA-based genotyping has advantages over tissue-based genotyping in a short turnaround time, invasiveness, and feasibility in serial assessment [233-235]. Due to the limitation of tissue-based genotyping, which can be affected by tissue accessibility or tumor purity, ctDNA-based genotyping can be conducted as initial genotyping in the rapidly growing aggressive tumor when challenges or delays in sample acquisition are anticipated. In addition, the ctDNA-based genotyping first approach can be preferred for the evaluation of emerged resistance mechanism [236]. ctDNA-based genotyping can also be used as a complementary method, either concurrently or sequentially with tissue-based genotyping in case of incomplete tumor genotyping or foreseen inadequate results due to uncertain adequacy of tissue [237].
Before genotyping ctDNA sequences, the concentration of cell-free DNA in plasma can be used as a prognostic biomarker [238,239]. The sensitivity of ctDNA assay varies among the primary sites and tumor types and should be considered at applying ctDNA test in clinical use [240]. Similarly, the metastatic site of the tumor affects the ctDNA detection and should be taken into account for using ctDNA assay [241]. Additionally, MSI-H/MMR-D and TMB-H, as determined by ctDNA assay, have been widely studied [242-244]. Improving the accuracy of the MSI detection and TMB calculation from ctDNA and defining reliable criteria for MSI-H/MMR-D and TMB-H in the ctDNA assay is anticipated to broaden the use of ctDNA tests.
CONCLUSION
NGS-based genetic testing has become an essential tool in treating patients with advanced solid cancers. This report provides clinical recommendations for the application of NGS in such patients, offering expert opinions on its diagnostic uses, and gene classification in accordance with K-CAT, while taking the domestic Korean context into consideration.
As cancer genomics advances and new therapies emerge, the current gene classification is subject to dynamic changes, and the application of NGS is anticipated to continuously evolve. Consequently, healthcare providers and researchers are encouraged to stay abreast of the latest advancements in the field of precision oncology to ensure optimal patient care and further cancer research.
Notes
Ethics Statement
Not applicable.
Availability of Data and Material
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: JHK, WSK, MK. Data curation: all authors. Writing—original draft: all authors. Writing—review & editing: all authors. Approval of final manuscript: all authors.
Conflicts of Interest
S.Y.P., the editor-in-chief and H.S.L. and S.O.Y., contributing editors of the Journal of Pathology and Translational Medicine, were not involved in the editorial evaluation or decision to publish this article. All remaining authors have declared no conflicts of interest.
Funding Statement
This study was supported by the National R&D Program for Cancer Control through the National Cancer Center (NCC) funded by the Ministry of Health & Welfare, Republic of Korea (HA22C0052).