Paricalcitol prevents MAPK pathway activation and inflammation in adriamycin-induced kidney injury in rats
Article information

Abstract
Background
Activation of the mitogen-activated protein kinase (MAPK) pathway induces uncontrolled cell proliferation in response to inflammatory stimuli. Adriamycin (ADR)-induced nephropathy (ADRN) in rats triggers MAPK activation and pro-inflammatory mechanisms by increasing cytokine secretion, similar to chronic kidney disease (CKD). Activation of the vitamin D receptor (VDR) plays a crucial role in suppressing the expression of inflammatory markers in the kidney and may contribute to reducing cellular proliferation. This study evaluated the effect of pre-treatment with paricalcitol on ADRN in renal inflammation mechanisms.
Methods
Male Sprague-Dawley rats were implanted with an osmotic minipump containing activated vitamin D (paricalcitol, Zemplar, 6 ng/day) or vehicle (NaCl 0.9%). Two days after implantation, ADR (Fauldoxo, 3.5 mg/kg) or vehicle (NaCl 0.9%) was injected. The rats were divided into four experimental groups: control, n = 6; paricalcitol, n = 6; ADR, n = 7 and, ADR + paricalcitol, n = 7.
Results
VDR activation was demonstrated by increased CYP24A1 in renal tissue. Paricalcitol prevented macrophage infiltration in the glomeruli, cortex, and outer medulla, prevented secretion of tumor necrosis factor-α, and interleukin-1β, increased arginase I and decreased arginase II tissue expressions, effects associated with attenuation of MAPK pathways, increased zonula occludens-1, and reduced cell proliferation associated with proliferating cell nuclear antigen expression. Paricalcitol treatment decreased the stromal cell-derived factor 1α/chemokine C-X-C receptor type 4/β-catenin pathway.
Conclusions
Paricalcitol plays a renoprotective role by modulating renal inflammation and cell proliferation. These results highlight potential targets for treating CKD.
Chronic kidney disease (CKD) is characterized by damage to renal endothelial cells and epithelial cells, resulting in tissue fibrosis and inflammation [1]. The loss of cell differentiation in CKD is directly linked to the interaction of cytokines, inflammatory factors, transcription factors, and cellular pathway activation [2]. Inflammation of kidney tissue contributes to the progression of the fibrotic process and CKD, regardless of its etiology [3]. The inflammatory response to epithelial and endothelial cell lesions involves the release of profibrotic and pro-inflammatory cytokines and chemokines, which induce the infiltration of inflammatory cells, mainly macrophages and monocytes, to the glomerulus and renal interstitium [4]. Molecules such as reactive oxygen species, tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β) are produced by M1 macrophages. During the repair phase, the macrophage phenotype is converted to M2, conferring the secretion of anti-inflammatory cytokines such as interleukin-10, which contributes to the resolution of the tissue injury [5].
Adriamycin-induced nephropathy (ADRN) is one of the most relevant rat models used to study the mechanisms involved in the pathogenesis of CKD. The ADRN rat model is similar to focal segmental glomerulosclerosis in humans, showing morphological characteristics of damage to a portion (segmental) of the glomerular capillaries in some of the glomurulus (focal area). These alterations result in renal dysfunction, glomerular fibrosis, and progressive albuminuria, which was seen in our previous work [1]. Studies have shown that tubulointerstitial inflammation in the ADRN model results in the recruitment of immune cells that secrete pro-inflammatory and profibrotic cytokines such as transforming growth factor β (TGF-β), TNF-α, and IL-1β [6,7]. The mitogen-activated protein kinase (MAPK) pathway also participates in adriamycin (ADR)-induced tubular injury to induce uncontrolled cell proliferation and differentiation in response to extracellular and intracellular stimuli [8]. Smad-dependent TGF-β is the main pathway in the pathogenesis of fibrosis and inflammation in CKD. Smad-independent pathways can also be activated to mediate renal fibrosis, such as the MAPK pathways mediated by extracellular signal-regulated kinase (ERK1/2), p38 MAPK and c-Jun N-terminal kinase (JNK), which represent non-canonical pathways activated by TGF-β1 [9].
Vitamin D receptor (VDR) activators have been used to evaluate the effects of vitamin D (Vit. D) in various experimental and clinical models of kidney disease [10]. Paricalcitol [19-nor-1.25 (OH2D2)], one of the synthetic analogs of calcitriol, exhibits immunomodulatory effects by inhibiting the secretion of proinflammatory cytokines [11,12] and profibrotic cytokines such as TGF-β, which consequently decreases cell loss and epithelial-mesenchymal transition by directly activating VDR [1,10]. Vit. D in its active form improved the inflammation and oxidative stress in acute kidney injury induced by cisplatin in rats [13] and exhibited protective effects on the outer and inner medullary structure in progressive kidney disease caused by a lack of renin-angiotensin system during rat kidney development [14]. Previous studies from our laboratory have shown that deficiency of Vit. D during adulthood in rats worsened renal function and disrupted renal structure when associated with diabetes [15], and its supplementation improved endothelial function and structure [5]. However, the direct effects of prior activation of the VDR before the onset of CKD have not yet been fully elucidated. These findings may contribute to better understanding of the mechanisms involved in preventing the progression of CKD. The present work evaluated the effect of paricalcitol treatment on the activity of inflammatory pathways in the ADR-induced CKD model.
MATERIALS AND METHODS
Animals and experimental design
The experimental protocol followed the ethical principles of animal experimentation outlined by the National Council for the Control of Animal Experimentation (CONCEA). This study was approved by the Ethics Committee on the Use of Animals (CEUA) of the Ribeirão Preto Medical School - University of São Paulo (FMRP-USP) (approval no. 194/2017).
Male Sprague-Dawley rats weighing 180–200 g were housed in a temperature-controlled environment (22°C) under a 12-hour light/dark cycle in the animal housing center of the Renal Physiology Laboratory. The animals received diet and water ad libitum. The animals were divided into four experimental groups: control: rats received 0.9% NaCl solution by pump and intravenous injection, n=5; paricalcitol: rats received paricalcitol (6 ng/day) by pump and 0.9% NaCl solution by intravenous injection, n=5; ADR: rats received 0.9% NaCl solution by pump and ADR by intravenous injection (3.5 mg/kg), n=7; and ADR+paricalcitol: rats received paricalcitol (6 ng/day) by pump and ADR by intravenous injection (3.5 mg/kg), n=7. Paricalcitol (6 ng/day, Zemplar, Abbvie Laboratories, Abbott Park, IL, USA) or vehicle (0.9% sodium chloride [NaCl] solution) was administered via a mini osmotic pump (Model 2004, Alzet, Cupertino, CA, USA) surgically implanted in the animal’s back under inhalation anesthesia with isoflurane (Cristália, Sao Paulo, Brazil). The infusion was continuous throughout the 27 days of observation. At 48 hours after implantation, the animals received ADR (3.5 mg/kg, doxorubicin hydrochloride/Fauldoxo, Libbs, Sao Paulo, Brazil) or vehicle (0.9% NaCl solution) via an intravenous injection in the tail vein.
The animals were followed up for 4 weeks after the single injection of ADR without any intervention. On the 27th day, the animals were anesthetized to remove the kidneys for immunohistochemistry, enzyme-linked immunosorbent assay (ELISA) and Western blot.
Immunohistochemical analysis
The kidney tissue was fixed with methacarn solution (60% methanol, 30% chloroform, and 10% acetic acid); after 24 hours, the solution was replaced with 70% alcohol. The tissue was embedded in paraffin, deparaffinized, and hydrated. Nonspecific antigen binding was blocked by incubation for 20 minutes with goat serum and endogenous peroxidase blocking. The slides were microwaved and immersed in citrate buffer pH 6.0 or ethylenediaminetetraacetic acid (EDTA) pH 8.5 for antigen retrieval. The sections were incubated with antibody against cluster of differentiation 68 (CD68, 1/100, MCA341R, Bio-Rad Laboratories Inc., Hercules, CA, USA) overnight at 4°C. The slides were then washed with buffer (phosphate bufered saline, 0.15 M NaCl, and PO4 buffer, pH=7.4) and incubated with anti-mouse secondary antibody for 30 minutes. Avidin-biotin-peroxidase complexes (Vector Laboratories, Newark, CA, USA) and 3,3'-diaminobenzidine (Sigma Chemical Company, St. Louis, MO, USA) were used to detect the antigens. The sections were counterstained with methyl green, dehydrated, and mounted. Images were obtained and data were quantified at high magnification (400×). Thirty consecutive 0.1 mm2 fields of the cortex and 20 consecutive 0.1 mm2 fields of the outer medullary compartment were evaluated for CD68 expression. Additionally, 30 cortical and 20 juxta-medullary glomeruli were assessed. The glomerular, tubulo-interstitial cortical, and medullary changes were quantified using NIH Image J software (Bethesda, MD, USA), and the average values per kidney were calculated. The results are expressed as the percentage of positive labeling in the glomerulus, cortex, and outer medulla.
ELISA analysis
Kidney tissues were homogenized in lysis buffer (50 mM Tris-hydrochloric acid [HCl], pH 7.4; 150 mM NaCl; 1% Triton X-100; protease inhibitor cocktail [100×] and 0.001 M EDTA, pH 8 [Thermo Fisher Scientific Inc., Waltham, MA, USA]) and centrifuged at 4°C at 10,000 rpm for 20 minutes. The concentrations of TNF-α and IL-1β in kidney tissue homogenates were evaluated by ELISA kits (R&D Systems Inc., Minneapolis, MN, USA). The Bradford method was used to determine the protein levels in lysates; the results were corrected for the total protein quantity in tissue and expressed in pg/mg of tissue protein.
Western blot analysis
Kidney tissues were lysed as for ELISA. Protein samples (60 μg) were heated to 100°C in sample buffer containing β-mercaptoethanol. The samples were separated by polyacrylamide gel electrophoresis (sodium dodecyl sulfate polyacrylamide gel electrophoresis, 12%) and transferred to nitrocellulose membranes overnight at 4ºC. The membranes were incubated for 1 hour in blocking buffer (Tris-buffered saline Tween 20, TBSt, 5% skimmed milk) or 3% bovine serum albumin and washed in buffer (TBSt, 0.1% Tween 20, pH 7.6). The membranes were then incubated overnight at 4°C with antibodies against arginase I (1/1,000, sc-18351, Santa Cruz Biotechnology, Santa Cruz, CA, USA), arginase II (1/1,000, sc-18357, Santa Cruz Biotechnology), CD68 (1/2,000, MCA341R, Bio-Rad Laboratories Inc.), C-X-C chemokine receptor type 4 (CXCR4; 1/500, LS-B6709, LifeSpan BioSciences Inc., Lynnwood, WA, USA), 24-hydroxylase (CYP24A1, 1/500, H0000159-M02, Abnova, San Diego, CA, USA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1/1,000, cod. 2118L, Cell Signaling Technology, Danvers, MA, USA), nuclear factor κB alpha inhibitor (IκBα, 1/1,000, sc-371, Santa Cruz Biotechnology), nuclear factor κB beta inhibitor (IκBβ, 1/1,000, sc-945, Santa Cruz Biotechnology), nuclear factor-κB (NF-κB; 1/1,000, sc-7151, Santa Cruz Biotechnology), proliferating cell nuclear antigen (PCNA; 1/500, P8825, Sigma Chemical Company), phospho-extracellular signal-regulated kinase 1/2 (p-ERK1/2; 1/500, sc-7383, Santa Cruz Biotechnology); phospho-c-Jun N-terminal kinase (p-JNK; sc-6254, Santa Cruz Biotechnology), phospho-p38 MAPK (1/500, P8825, Sigma Chemical Company), stromal cell-derived factor 1α (SDF-1α; 1/500, 14-7992-83, eBioscience, San Diego, CA, USA), zonula occludens-1 (ZO-1; 1/250, 61-7300, Zymed, Carlsbad, CA, USA), and β-catenin (1/2,000, sc-7199, Santa Cruz Biotechnology). The membranes were then washed and incubated with peroxidase-conjugated mouse anti-IgG (P0447, 1/5,000, Dako Corporation, Copenhagen, Denmark), rabbit anti-IgG (P0448, 1/2,000, 1/5,000 or 1/10,000, Dako Corporation or sc-2357 [Santa Cruz Biotechnology]) or anti-goat (sc-2768, 1/5,000, Santa Cruz Biotechnology) for 1 hour at room temperature. GAPDH was used as the loading control. Bands were visualized using enhanced chemiluminescence reagents (Sigma-Aldrich) and an imaging system (Kodak Gel Logic 2200, Austin, TX, USA). Band intensity was quantified by densitometry using ImageJ NIH 1.52A imaging software (http://www.nih.gov). Protein expression was determined as the percentage of the band density of the protein of interest and the reference protein compared with the control group. The control value was designated as 100%.
Statistical analysis
Data were tested for normality using the Kolmogorov-Smirnov distribution normality test. One-way analysis of variance (ANOVA) followed by the Newman-Keuls multiple comparisons test was used to analyze normally distributed data. Data are expressed as mean±standard error of the mean. The Kruskal-Wallis nonparametric test, followed by Dunn’s post-test, was used to analyze non-normally distributed data expressed as medians and percentiles (25%–75%). Statistical analyses were conducted using GraphPad Prism ver. 9.0 for Windows (GraphPad Software, San Diego, CA, USA). p<.05 was considered statistically significant.
RESULTS
Paricalcitol treatment increased Vit. D content in kidney tissue
The expression of CYP24A1, an enzyme that finely regulates Vit. D levels, was used as an indicator of activated intrarenal Vit. D content. A reduction of CYP24A1 expression was observed in the rats in the ADR group compared with the control and paricalcitol groups (Fig. 1), indicating less Vit. D in this tissue. These changes in CYP24A1 expression were significantly prevented in the ADR+paricalcitol group compared with the ADR group, which shows a higher content of the Vit. D in the kidneys of animals treated with paricalcitol.
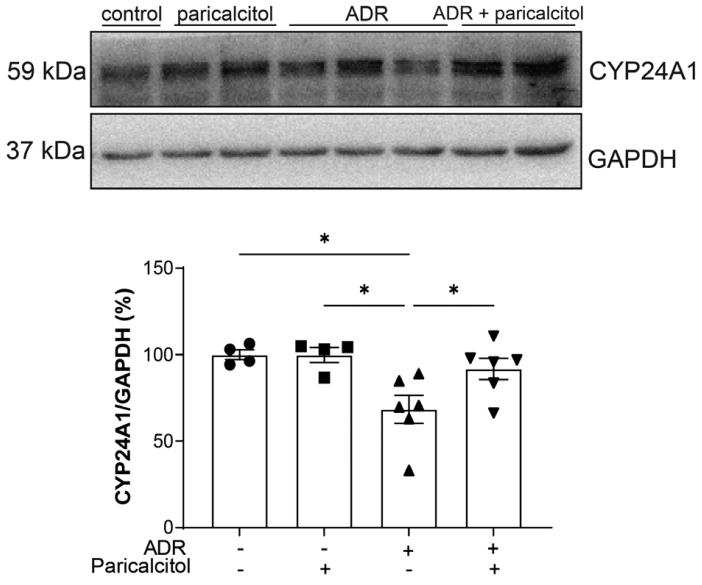
Western blot analysis of CYP24A1 in kidney. Glyceraldehyde-3-phosphate dehydrogenase (GADPH) was used as loading control. Data from the control (dots), paricalcitol (squares), adriamycin (ADR; upward facing triangles) and ADR + paricalcitol (downward facing triangles) groups. n = 4–6 for each group. One-way ANOVA and Newman-Keuls multiple comparisons; data are expressed as mean ± standard error of the mean. *p < .05.
Paricalcitol treatment attenuated macrophage infiltration induced by ADR
The expression of CD68, a marker of macrophages and monocytes, was increased in the glomerulus, cortex, and outer medulla of animals in the ADR group compared with the control and paricalcitol groups. Compared with the ADR group, the ADR+paricalcitol group showed a decrease in the expression of CD68 in all compartments (Fig. 2A–D). Western blot analysis of CD68 showed similar results (Fig. 2E).
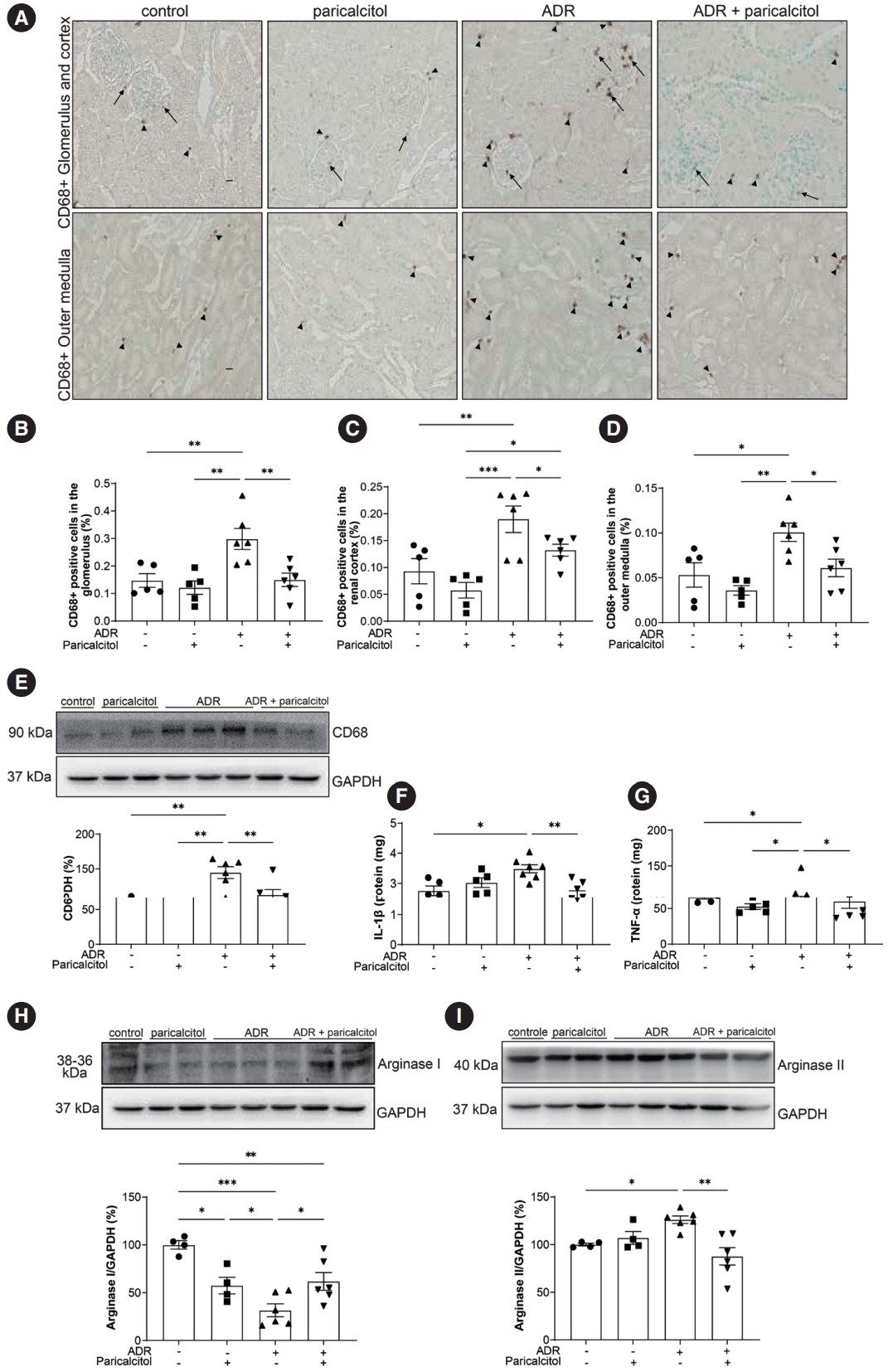
Analysis of macrophages in kidney tissue. (A) Immunolocalization of CD68 in renal compartments. Arrows indicate positive expression of CD68 in the glomerulus. Arrowheads indicate positive CD68 expression in the tubulointerstitial compartments. Percentage of CD68-positivity in the glomerulus (B), in the cortex (C), and in the outer medulla (D). (E) Densitometric analysis of CD68. Cytokines and macrophage profile in kidney tissue. Enzyme-linked immunosorbent assay for interleukin-1β (IL-1β) (F) and tumor necrosis factor-α (TNF-α) (G). Densitometric analysis of arginase I (H) and arginase II (I) in the kidney. Glyceraldehyde-3-phosphate dehydrogenase (GADPH) was used as loading control. Data from the control (dots), paricalcitol (squares), adriamycin (ADR; upward facing triangles) and ADR + paricalcitol (downward facing triangles) groups. n = 4–6 for each group. One-way ANOVA and Newman-Keuls multiple comparisons; data are expressed as mean±standard error of the mean. *p < .05, **p < .01, ***p < .001.
There was an increase in TNF-α and IL-1β (pro-inflammatory cytokines) in the ADR group compared with the control and paricalcitol groups. The cytokine levels were lower in the ADR+paricalcitol group than in the ADR group (Fig. 2F, G). A reduction in arginase I expression, here used as a repair macrophage marker, was observed in the kidney tissue of the animals in the ADR group compared with control and paricalcitol groups. This reduction was reversed in the ADR+paricalcitol group compared with the ADR groups. Also for arginase I, a lower expression was seen in the animals in the paricalcitol group, demonstrating the effect of this Vit. D analog in modulating the differentiation of tissue macrophages (Fig. 2H). Arginase II was increased in the ADR group compared with the control group. This increase was attenuated in the ADR+paricalcitol group compared with the ADR group (Fig. 2I).
Paricalcitol treatment regulated MAPK pathway activation and attenuated the changes in ZO-1 and PCNA expression induced by ADR
We evaluated the activation of MAPK-related pathways and the inflammatory process. The level of p-p38 was lower in the ADR+paricalcitol group compared with the control, paricalcitol, and ADR groups (Fig. 3A). An increase in p-JNK and p-ERK1/2 was seen in the ADR group compared with the control group; this increase was attenuated in the ADR+paricalcitol group (Fig. 3B, C).
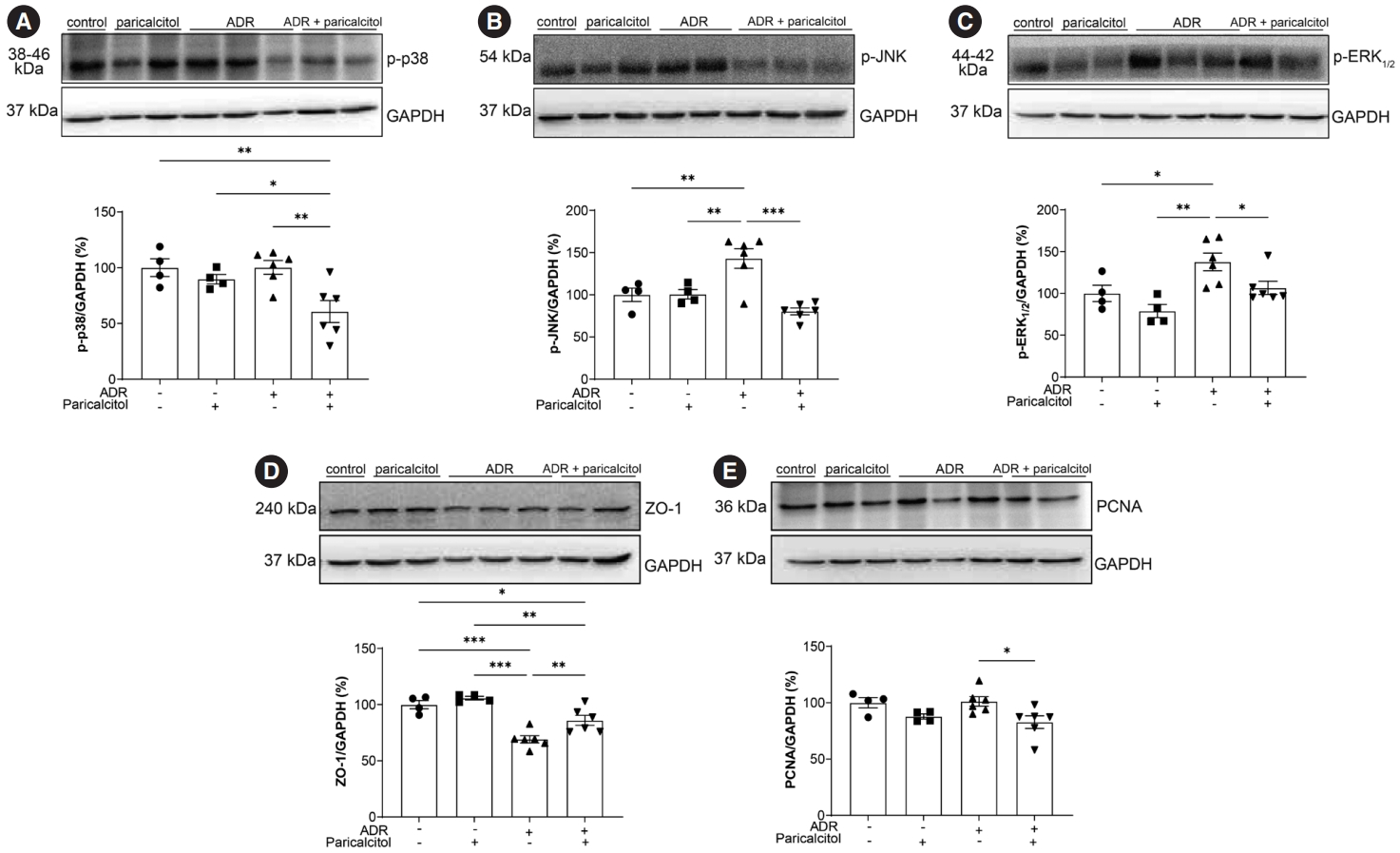
Activation of mitogen-activated protein kinase pathways. Analysis of p-p38 (A), p-JNK (B), p-ERK1/2 (C), zonula occludens-1 (ZO-1) (D), and proliferating cell nuclear antigen (PCNA) (E). Glyceraldehyde-3-phosphate dehydrogenase (GADPH) was used as loading control. Data from the control (dots), paricalcitol (squares), adriamycin (ADR; upward facing triangles) and ADR + paricalcitol (downward facing triangles) groups. n = 4–6 for each group. For (A–D), one-way ANOVA and Newman-Keuls multiple comparisons; data are expressed as mean±standard error of the mean. *p < .05, **p < .01, ***p < .001.
The expression of ZO-1 was decreased in the ADR group compared with the control and paricalcitol groups. The ADR+paricalcitol group also showed a reduction of ZO-1 expression compared to control and paricalcitol groups. However, this expression was increased when compared to ADR group (Fig. 3D). A significant reduction in PCNA in kidney tissue was observed in the ADR+paricalcitol group compared with the ADR group (Fig. 3E).
Paricalcitol treatment attenuated the pro-inflammatory pathways induced by ADR
No difference was observed in the expression of the NF-κB protein among the four groups (p>.05) (Fig. 4A). However, a reduction in the inhibitory proteins IκBα and IκBβ was observed in the ADR group compared with the control and paricalcitol groups. The ADR+paricalcitol group also significantly reduced IκBα levels compared to the control and paricalcitol groups, but increased compared to the ADR group (Fig. 4B). The same profile was observed for IκBβ (Fig. 4C).
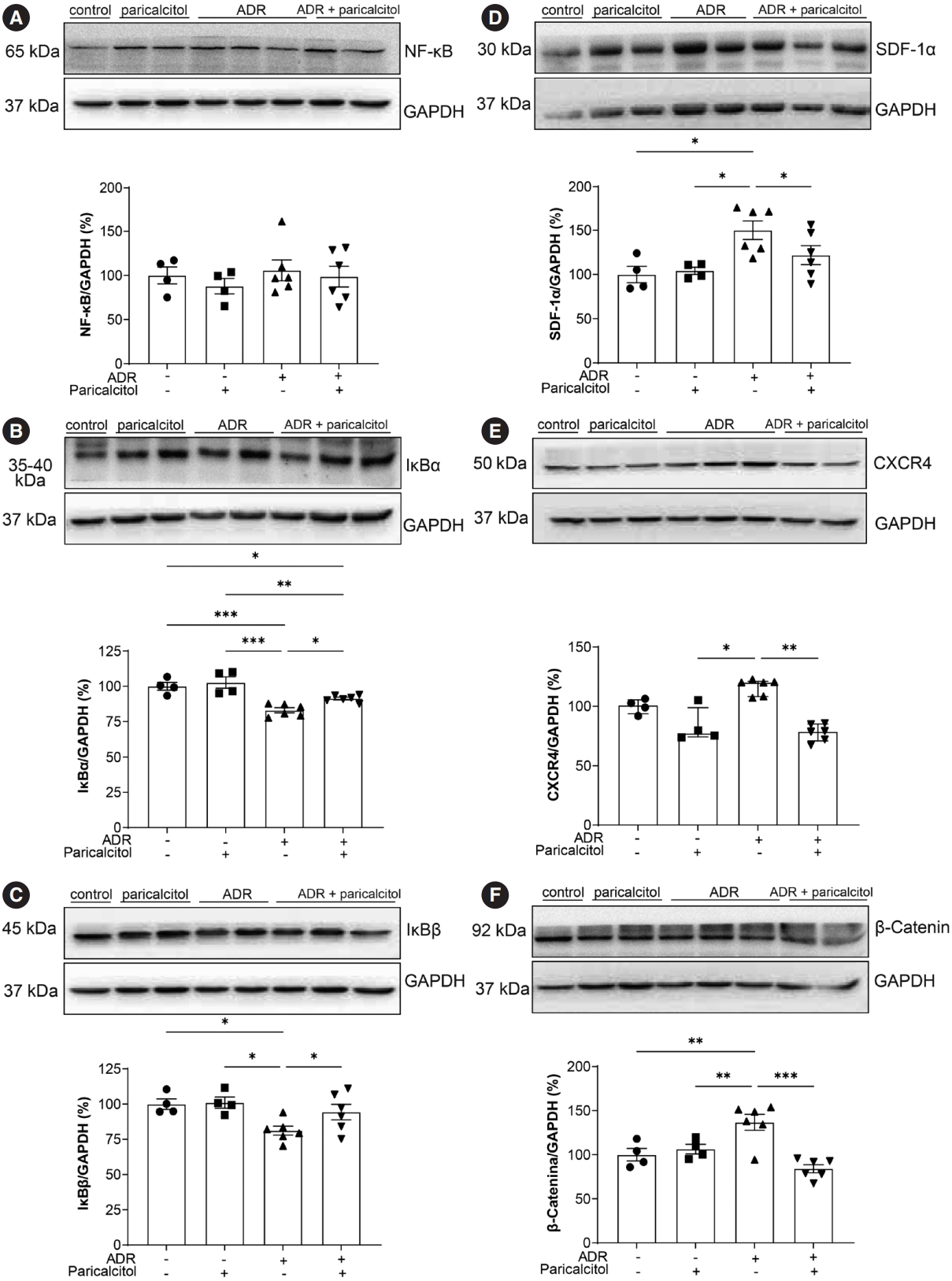
Analysis of pro-inflammatory pathways in renal tissue. Analysis of nuclear factor-κB (NF-κB) (A), nuclear factor κB alpha inhibitor (IκBα) (B), nuclear factor κB beta inhibitor (IκBβ) (C), stromal cell-derived factor 1α (SDF-1α) (D), C-X-C chemokine receptor type 4 (CXCR4) (E), and β-catenin (F). Glyceraldehyde-3-phosphate dehydrogenase (GADPH) was used as loding control. Data from the control (dots), paricalcitol (squares), adriamycin (ADR; upward facing triangles) and ADR + paricalcitol (downward facing triangles) groups. n = 4–6 for each group for (A, B, C, D, F). One-way ANOVA and Newman-Keuls multiple comparisons; data are expressed as mean ± standard error of the mean. (E) Kruskal-Wallis and Dunn’s post-test; data are expressed as median and percentiles (25%–75%). *p < .05, **p < .01, ***p < .001.
The SDF-1α/CXCR4 pathway was significantly increased in the ADR group compared with the control and paricalcitol groups. Treatment with active Vit. D attenuated the expression of these pro-inflammatory proteins (Fig. 4D, E). The β-catenin protein showed similar expression in the ADR group compared with the control groups and its expression was attenuated in the kidneys in the ADR+paricalcitol group (Fig. 4F).
DISCUSSION
In the present study, we investigated the role of active Vit. D in ADR-induced kidney damage. The protective effects of paricalcitol may be related to increased expression of CYP24A1, which contributed to reduced expression of mediators of the Smad2/3-independent TGF-β pathway, thus reducing the fibrotic process observed in our study. The data also demonstrated the effect of paricalcitol in reducing the inflammatory process in the ADR rat model. Together, these results demonstrate that paricalcitol may attenuate CKD by increasing the availability of active Vit. D in renal tissue to generate anti-inflammatory and anti-fibrotic responses. Vit. D levels are finely regulated by the CYP24A1 enzyme or 24-hydroxylase, which catabolizes calcidiol and calcitriol. Thus, CYP24A1 plays an essential role in modulating local Vit. D activity and can be an indicator of active Vit. D in tissue [16,17]. Vit. D in its active and circulating form may increase the levels of its receptor, VDR, as observed in our previous study [1], in ADR-induced kidney injury, contributing to the recovery of the changes in kidney function and structure.
Some evidence has indicated a therapeutic effect of Vit. D in CKD in humans, mainly due to its antifibrotic and anti-inflammatory effects related to the reduction of cytokine secretion via the VDR. Paricalcitol was also shown to reduce vascular calcification and help in the preservation of kidney function from its antiproteinuric effects [12,18]. The accumulation and migration of macrophages in kidney diseases are associated with functional and structural kidney damage [18]. Our study demonstrated that paricalcitol attenuated the accumulation of macrophages in all the compartments evaluated in the ADR+paricalcitol group animals and reduced the secretion of cytokines and activating pro-inflammatory pathways. TNF-α and IL-1β were increased in ADR animals, pointing to the response of pro-inflammatory activity caused by ADR in rats. In our study, paricalcitol likely exerted anti-inflammatory effects by increasing arginase I levels. Arginase I and II are involved in the production of NO by the endothelium and also with the immune system modulation. While arginase II overexpression in macrophages increases the production of pro-inflammatory cytokines, arginase I inhibits and attenuates inflammation in cardiac tissue [19]. In this study, we observed an increase in arginase I and a reduction in arginase II in the ADR animals treated with paricalcitol. These results demonstrated that paricalcitol has an anti-inflammatory action, as evidenced by a decrease in M1 macrophages and an increase in M2 macrophages. Thus, the macrophages in the kidneys of ADR+paricalcitol animals, at least part of them, have anti-inflammatory and reparative activity in the tissue, improving tissue fibrosis and modulating the immune response. Our study showed a reduction of arginase I in the paricalcitol group compared to control. This result reinforces the role of Vit. D in protecting tissue when an inflammatory process occurs. Otherwise, repair macrophages are not needed. Under normal conditions, macrophages are not recruited to the healthy tissue, with fewer pro-inflammatory and fibrotic actions induced by ADR and angiopoietin-2 [1].
ADR induces the phosphorylation of MAPKs (p38, JNK, and ERK1/2) [8], which may be associated with podocyte loss and proteinuria. MAPK signaling is triggered by mechanical and chemical stressors, leading to the regulation of cell proliferation, differentiation, and apoptosis [20,21]. We saw that all MAPK signaling pathways were activated in the kidneys of the ADR group compared with controls. Paricalcitol attenuated these changes, which contributed to the improvement in kidney function and structure. Vit. D plays an active regulatory role in attenuating the phosphorylation of ERK1/2, JNK and p38, which is reflected in a reduction in cell proliferation [22,23].
The progression of CKD is also associated with damage to structural proteins, such as ZO-1, which tightens epithelial cell layer junctions to create a selective paracellular barrier in the glomerulus and tubules [24]. ZO-1 is a cytoplasmic protein that transmits information to the nucleus in response to cell-cell contact. ZO-1 modulates cell cycle progression and controls cell proliferation and differentiation through interaction with ZO1-associated nucleic acid (ZONAB) [24]. ZONAB promotes the expression of the PCNA gene, promoting the cell cycle transition from G1 phase to S phase [25]. In our study, ZO-1 was decreased in the ADR group. The loss of cellular integrity and ZO-1 decrease can generate abnormal ZONAB transcripts, leaving this protein free to induce PCNA transcription [26]. There was an attenuation of PCNA in the kidney tissue of animals in the ADR+paricalcitol group. We also found that PCNA is attenuated by Vit. D, reducing the exacerbated proliferation of various cell types, especially mesenchymal cells and macrophages [13,15,23].
VDR activation also suppresses activation of the MAPKs and NF-κB pathways [27,28]. In our study, no differences were observed in the expression of NF-κB in the treated groups, although its two inhibitory proteins, IκBα and IκBβ, were decreased in the ADR+paricalcitol group. Studies have shown the anti-inflammatory role of active Vit. D in reducing the activation of TGF-β1/Smad2/3, a mechanism that contributes to the greater availability of Smad7, increasing the expression and activity of IκBα and contributing to the reduction of inflammatory cell infiltration in renal tissue [29]. Thus, these results suggest that Vit. D functions in these pathways, even if indirectly. In the ADRN model, CXCR4 is activated by the SDF-1α ligand, which triggers an increase in β-catenin to induce podocyte injury and proteinuria [30], showing new TGF-β–independent pathways for the evolution of proteinuric diseases. Previous study from our laboratory showed decreased CXCR4 in animals with acute kidney injury induced by cisplatin treated with calcitriol, with more significant regeneration of tubular cells and less endothelial dysfunction [13]. Active Vit. D reduces the expression of profibrotic pathways such as Wnt/β-catenin, decreasing glycogen synthase kinase-3β and Snail, proteins involved in the epithelial-mesenchymal transition. Decreasing this mechanism also leads to lower proteinuria through the direct physical interaction of VDR/β-catenin, reducing nuclear β-catenin and therefore cell proliferation [31]. Again, in our model, we observed the direct and indirect actions of paricalcitol and endothelium-tubular cell interaction to reduce mechanisms of inflammation and fibrosis in CKD.
Our study showed that paricalcitol attenuates the renal inflammatory process induced by ADR in rats. Paricalcitol induced anti-inflammatory and anti-proliferative effects by inhibiting macrophage accumulation and cytokine secretion and inducing M2 macrophages in renal tissue, regulating the MAPK, NF-κB/IκBα/IκBβ and CXCR4/SDF-1α/β-catenin pathways. Our results suggest that paricalcitol may exhibit anti-fibrotic, anti-inflammatory, and anti-proliferative therapeutic roles in preventing renal injury in early CKD.
Notes
Ethics Statement
The animal study protocol was approved by the Brazilian College of Animal Experimentation and the Animal Experimentation Committee of the University of Sao Paulo at Ribeirao Preto Medical School (COBEA/CETEA/FMRP-USP, protocol no. 194/2017).
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: ALD, LFA, BMO, CSS, ALDM, HDCF, CG, RSC, TMC. Data curation: ALD, BMO, CSS, ALDM. Formal analysis: ALD, BMO, CSS, ALDM. Funding acquisition: TMC, RSC. Methodology: HDCF, CG. Resources: TMC, RSC. Writing—original draft: ALD, LFA. Writing—review & editing: ALD, LFA, BM, CSS, ALDM, HDCF, CG, RSC, TMC. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
This work was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superi-or – Brasil (CAPES) – Finance Code 001 and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grant number 303252/2021-9).
Acknowledgements
The authors thank Guilherme Lemos and Flávio Leite for help with the immunohistochemistry sections.