Multifocal Renal Cell Carcinoma of Different Histological Subtypes in Autosomal Dominant Polycystic Kidney Disease
Article information
Abstract
Renal cell carcinoma (RCC) in autosomal dominant polycystic kidney (ADPKD) is rare. To date, 54 cases of RCC in ADPKD have been reported. Among these, only 2 cases have different histologic types of RCC. Here we describe a 45-year-old man who received radical nephrectomy for multifocal RCC with synchronous papillary and clear cell histology in ADPKD and chronic renal failure under regular hemodialysis. The case reported herein is another example of the rare pathological finding of RCC arising in a patient with ADPKD.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common hereditary renal cystic disease. The development of renal cell carcinoma (RCC) is one of the most serious complications of ADPKD. The importance of careful monitoring for malignant transformation has been emphasized in patients with ADPKD. However, the clinical diagnosis of RCC in patients with ADPKD is difficult because it has no specific symptoms and underlying distortion of the renal architecture caused by ADPKD disturbs radiologic detection.1
To date, 54 cases of RCC associated with ADPKD have been reported. In these cases, clear cell RCC was the most common histological subtype (48%), followed by papillary RCC (14%).1-13 There were only two cases with the coexistence of different histological subtypes of RCC.3,5 Here, we report a case of multifocal RCC with synchronous papillary and clear cell histology occurring in a patient with ADPKD.
CASE REPORT
A 45-year-old man was admitted for the evaluation of recurrent abdominal pain. He had a 10-year history of hemodialysis for chronic renal failure due to ADPKD. His symptoms developed recently, and he denied having a fever, chronic fatigue, hematuria, or weight loss. The patient's laboratory results were significant for proteinuria (2,860 mg/day). On computed tomography (CT), both kidneys had multiple thin-walled cysts, and a heterogeneous well-enhanced mass was present in the mid-pole of the right kidney (Fig. 1A). Neither enlarged lymph nodes nor renal vein thrombosis was noted. A right radical nephrectomy was performed with the presumptive clinical diagnosis of RCC.
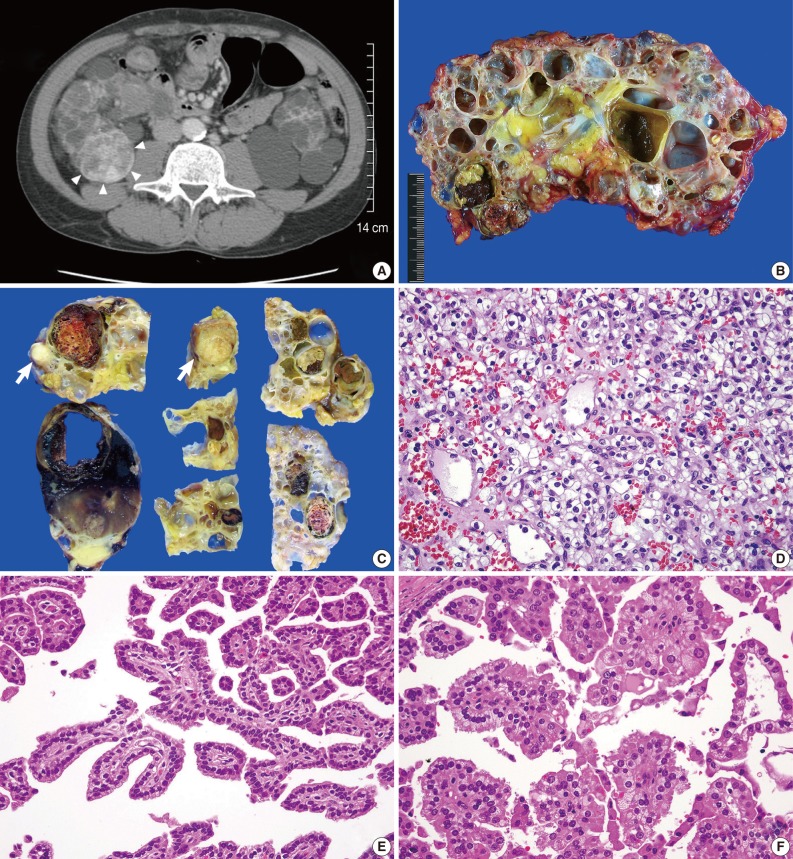
(A) Abdominal computed tomography shows a heterogeneous, well-enhanced mass in the mid-pole of the right kidney (arrowheads). The bilateral renal parenchyma is replaced by multiple, thin-walled cysts. (B) Numerous, variable-sized cysts occupy most of the renal parenchyma. A few solid masses are also identified between the cysts. (C) Fifteen of the 17 solid tumor masses detected during serial section show a well-circumscribed, round to ovoid, variegated appearance, with frequent areas of necrosis and hemorrhage. In contrast, the remaining two masses indicated by arrows display a distinctive gross feature characterized by a whitish tan, solid or granular cut surface without necrosis or hemorrhage. (D) Histologically, clear cell renal-cell carcinoma (RCC) is characterized by an alveolar growth pattern with frequent vascular proliferation. The tumor cells have a round or polygonal shape and clear to eosinophilic granular cytoplasm. (E, F) Papillary RCCs are characterized by a single layer of tumor cells with small nuclei, inconspicuous nucleoli, and pale to basophilic cytoplasm (type 1 papillary RCC; E), and pseudostratified tumor cells with large nuclei, prominent nucleoli, and eosinophilic cytoplasm (type 2 papillary RCC; F).
The resected kidney measured 16×10×10 cm and showed numerous, variable-sized, thin-walled cysts that occupied almost the entire parenchyma (Fig. 1B). In addition to the radiologically detected mass, which measured 6×4×4 cm, 16 more solid tumor masses were scattered throughout the kidney from the upper pole to the lower pole. The sizes of these 16 tumor masses ranged from 0.5×0.4×0.4 cm to 2.8×2.5×2.1 cm. All masses were confined to renal parenchyma without penetrating the renal capsule and they did not invade the renal veins. Fifteen of the 17 masses were well-circumscribed but unencapsulated, round to ovoid, variegated in appearance, and with frequent areas of necrosis and hemorrhage (Fig. 1C). These 15 masses had typical histological features of clear cell RCC characterized by a solid to alveolar growth pattern with rich vascular networks and occasional tubule formation. The tumor cells were round or polygonal and had cytoplasm that varied from clear to eosinophilic granular (Fig. 1D). Fuhrman's grade for most of the masses was 2, but in a few areas was grade 4. In contrast, the other two masses were well-circumscribed and encapsulated, globular in appearance with a whitish tan, granular cut surface (Fig. 1C). The masses showed typical histological features of papillary RCC, characterized by a single layer or pseudostratified layers of tumor cells arranged on fibrovascular stalks. Both tumors consisted of a mixture of two subtypes of papillary RCC: a single layer of tumor cells with small nuclei, inconspicuous nucleoli, and pale to basophilic cytoplasm (type 1 papillary RCC) (Fig. 1E); and pseudostratified tumor cells with large nuclei, prominent nucleoli, and eosinophilic cytoplasm (type 2 papillary RCC) (Fig. 1F). The proportion of type 1 papillary RCC was slightly greater than that of type 2 papillary RCC. Almost all cysts occupying the renal parenchyma were lined by a single layer of cuboidal to flat epithelium. A few cysts were lined with 3-6 layers of cuboidal epithelium. The residual renal parenchyma showed atrophic tubules and varying degrees of chronic inflammation, fibrosis, and hemosiderin pigmentation.
The patient was discharged 7 days after surgery without any complications. He has been followed as an outpatient and is doing well at 6 months without any signs of tumor recurrence.
DISCUSSION
To our knowledge, only 54 cases of RCC arising in patients with ADPKD have been reported.1-13 The association between RCC and chronic renal failure in patients with ADPKD remains controversial. In a review of 25 patients with ADPKD associated RCC by Keith et al.,11 4 patients were in chronic renal failure and 2 of these 4 patients were receiving dialysis. The authors suggested that there is no clear evidence for an association between RCC and chronic renal failure in patients with ADPKD.11 However, RCC has recently been found in patients with ADPKD and chronic renal failure in a large population-based study. Hajj et al.3 found RCC in 10 of 79 (12%) patients with ADPKD and chronic renal failure, and, among these, seven had been on dialysis for 1-13 years, and two had kidney transplants. Nishimura et al.1 demonstrated RCC in 10 of 510 (2%) patients with ADPKD and indicated that these 10 patients had been receiving dialysis for 5-17 years. Upon review of 9 case reports and our case, we found that 9 of 10 (90%) patients had chronic renal failure and received dialysis for 8-15 years.2,4-10,12,13
Including our patient, 33 patients with ADPKD associated RCC have been found to be in chronic renal failure among 55 patients with ADPKD associated RCC. Although there is no definitive evidence for an association between RCC and the progression to chronic renal failure in patients with ADPKD, these findings suggest that careful observation is warranted in patients with ADPKD and chronic renal failure. In our case, no abdominal CT scans of the patient were performed during 10 years of hemodialysis because he did not complain of any symptoms. Regular CT scanning combined with clinical follow-up could be helpful for early detection of RCC.
Keith et al.11 suggested that ADPKD-associated RCC has distinct clinical and pathological features, including development at a younger age, frequent bilaterality and multifocality, and higher frequency of the sarcomatoid type. Of the 55 patients who had ADPKD associated RCC including our patient, 14 (25%) showed multifocality and 16 (29%) showed bilaterality.1-14 Multifocality and bilaterality are significantly higher in patients with RCC and ADPKD than in those with sporadic RCC (25% and 29% vs 3% and 4%, respectively). Furthermore, seven of 14 (50%) multifocal RCCs found in patients with ADPKD showed bilaterality (Table 1).1,3,5,11 In our case, multiple small masses in the right kidney were not detected by abdominal CT, and there was a possibility that the left kidney also had small RCCs. However, a contralateral nephrectomy was not performed for reasons of co-morbidity.
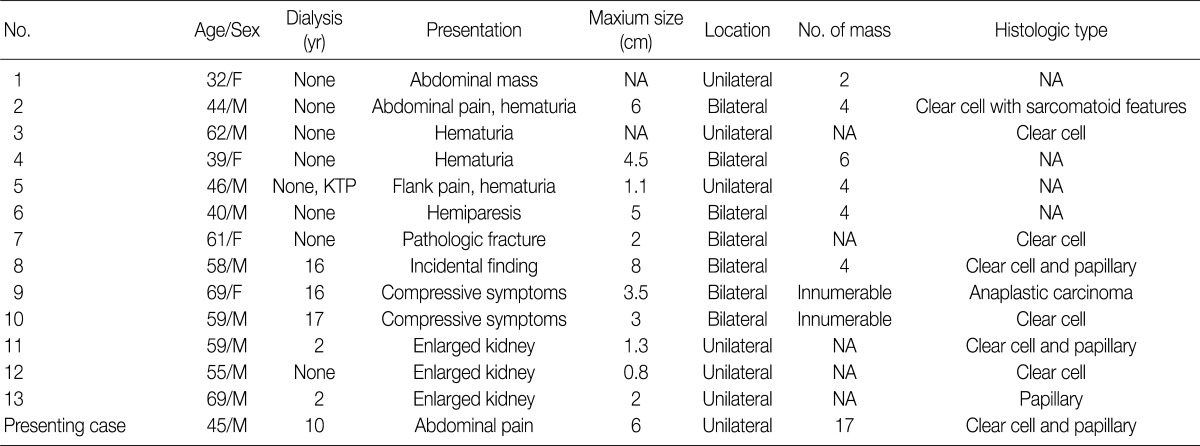
The histological type of RCC has been documented in 42 cases. Histologically, 25 of 42 cases (60%) showed clear cell RCC and 6 of 42 cases (14%) showed papillary RCC.1-14 Although Hajj et al.3 found a high frequency of papillary RCC compared to sporadic RCC in their study, the frequency of papillary RCC is similar in patients with ADPKD-associated RCC and those with sporadic RCC (14% vs 12%).14 In contrast, the combination of the two different histological types was seven times higher in patients with ADPKD-associated RCC compared to those with sporadic RCC (7% vs 1%).3,5,14
Some observations suggest that ADPKD affects the development of RCC. First, epithelial hyperplasia is frequently identified in patients with ADPKD. Atypical cells found in epithelial hyperplasia have been hypothesized to represent early neoplastic lesions in patients with acquired cystic kidney disease. Second, papillary adenoma, which is considered a precursor of papillary RCC, is frequent in patients with ADPKD.15 In contrast, some authors have explained ADPKD-associated RCC by simple coincidence because there is no definite evidence that RCC more frequently develops in patients with ADPKD.3,11 Others have suggested that ADPKD-associated RCC is a result of chronic kidney injury caused by ADPKD.1,8,13
Currently reported cases show that RCC is found in a large proportion patients with chronic renal failure in their 50s or 60s.1-14 Although the pathogenesis of RCC in patients with ADPKD is still debatable, chronic kidney injury may play a role in the development of RCC. Frequent multifocality and bilaterality may be evidence to support this idea. It is well known that long-standing inflammatory bowel disease causes malignant transformation that shows frequent multifocality.16 Similarly, acquired-renal cystic disease associated RCC which develops after several years of dialysis, shows frequent multifocality and bilaterality.15 Because almost the entire renal parenchyma undergoes chronic inflammation in patients with ADPKD, many cells are prone to genetic mutation leading to malignant transformation.
In conclusion, evidence is increasing that RCC is found in patients with ADPKD and chronic renal failure. Considering that ADPKD is a common cause of chronic renal failure, careful observation for the possible development of RCC should be mandatory in patients with ADPKD. Furthermore, recently reported cases demonstrated the aggressive behavior of RCC in ADPKD, which is characterized by frequent multifocality and bilaterality. The case reported herein is another example of the rare pathological finding of RCC arising in a patient with ADPKD.
Notes
No potential conflict of interest relevant to this article was reported.