Methylation and Immunoexpression of p16INK4a Tumor Suppressor Gene in Primary Breast Cancer Tissue and Their Quantitative p16INK4a Hypermethylation in Plasma by Real-Time PCR
Article information
Abstract
Background
The p16INK4a gene methylation has been reported to be a major tumorigenic mechanism.
Methods
We evaluated the methylation status of the p16INK4a genes in 231 invasive breast cancer and 90 intraductal carcinoma specimens using a methylation-specific polymerase chain reaction and p16 protein expression using immunohistochemistry. The quantity of cell-free methylated p16INK4a DNA in the plasma samples of 200 patients with invasive breast cancer was also examined using a fluorescence-based real-time polymerase chain reaction assay.
Results
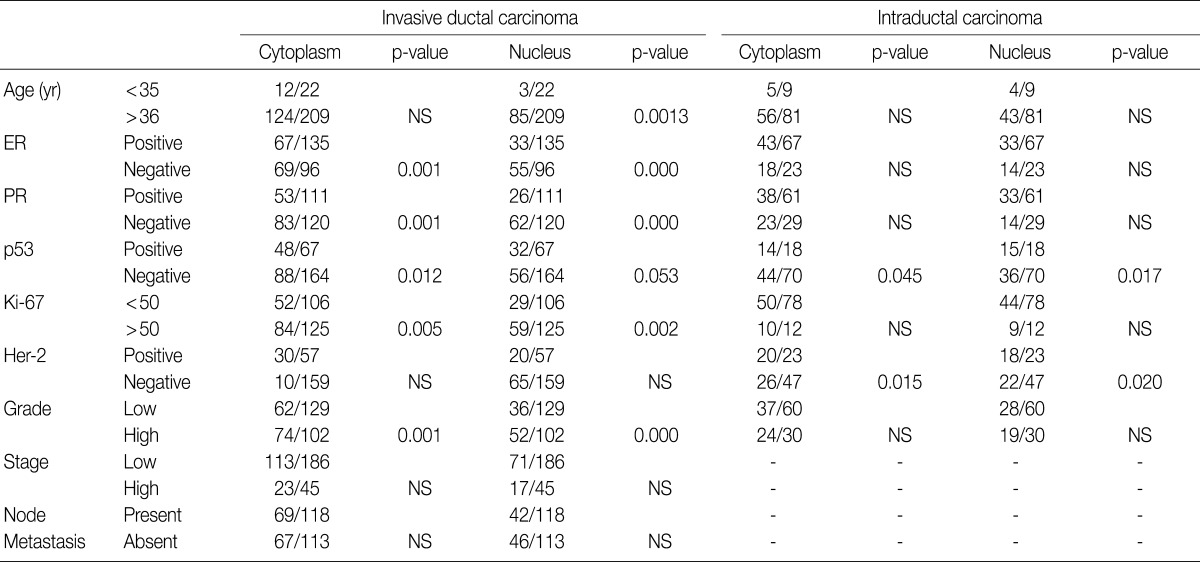
The frequencies of p16INK4a methylation in invasive and intraductal tumors were 52.8% (122/231) and 57.8% (52/90), respectively. The p16 protein was overexpressed in 145 of the 231 invasive carcinomas (62.8%) and 63 of the 90 intraductal carcinomas (70%). High p16 expression in invasive carcinomas correlated significantly with a high histologic grade, a negative estrogen receptor and progesterone receptor status, p53 immunoreactivity and high Ki-67 expression with immunohistochemistry. In addition, the methylation index of p16INK4a was significantly higher in the cancer patients than the normal controls (p<0.001).
Conclusions
High p16 immunoreactivity correlated with a loss of differentiation in breast carcinomas and high frequency of p16INK4a promoter methylation in both invasive and intraductal carcinomas, suggesting it may be involved in the pathogenesis of breast cancer.
The p16 protein is an inhibitor of cyclin-dependent kinase that blocks the G1/S phase of the cell cycle by inhibiting cyclin D-CDK4/6 complex formation through direct binding with cyclin dependent kinase (CDK) 4/6.1 The p16-CDK4/6 complex inhibits pRb phosphorylation, resulting in G1 arrest.2 The tumor suppressor p16INK4a gene is a major target in carcinogenesis, and down-regulation of the p16 protein has been reported in many malignancies.3 Expression of p16 has been reported in benign breast lesions such as fibroadenoma and in breast carcinoma.4 Some reports have also indicated that strong p16 expression is associated with poor prognostic parameters.5-7 Therefore, the pattern of p16 expression is variable and it complicates the elucidation of the role of p16INK4a in breast tumors.
Similar to many other genes, p16INK4a is commonly inactivated by hypermethylation of its CpG-rich promoter regions.3 p16INK4a promoter methylation has been detected in breast carcinoma, although the reported prevalence is discordant among different studies.3 Methylation of p16INK4a has been found in many precancerous lesions, such as dysplasia of the oral cavity, non-neoplastic mucosa near stomach cancer, ulcerative colitis, and the bronchial epithelium of chronic smokers without evidence of malignancy.8-10 The possibility has been raised of using DNA methylation as a tumor marker for cancer screening and treatment monitoring. Several groups have found methylated genes in the plasma of patients with cancers of the head and neck, esophagus, stomach, colon, rectum, breast, lung, and liver.11-17 Therefore cancer-specific methylation of DNA might be considered as a tumor marker and may be a good screening method for early cancer detection in high-risk groups and for the early detection of subclinical residual tumors after cancer treatment. Moreover, because such methylated promoter DNA that is detected in circulating blood is derived mainly from cancer cells, we hypothesized that the amount of methylated promoter DNA in the blood of cancer patients would be higher than that in healthy individuals and this increase may represent a possible screening marker.
In this study, we assessed p16 aberrant gene expression and p16INK4a methylation, and correlated these with the clinicopathologic parameters in breast cancer. In addition, the amount of methylated p16INK4a DNA in the plasma of patients with invasive breast cancer and of the normal controls was determined using a real-time fluorogenic polymerase chain reaction (PCR) approach.
MATERIALS AND METHODS
Study material and DNA extraction
We obtained tumor specimens from 231 patients with invasive breast carcinoma and from 200 matched preoperative plasma and tumor specimens from 90 patients with intraductal carcinoma who underwent surgical resection at Samsung Medical Center from May 2002 through December 2004. All of the tissues had been fixed in 10% neutral buffered formalin and embedded in paraffin and were evaluated for a wide variety of pathologic features by examination of the hematoxylin and eosin (H&E)-stained tissue sections and the immunohistochemically-stained slides by two investigators in a blinded fashion without knowledge of the molecular data. The medical records were reviewed for clinical information. The tumor grade and stage were determined based on the American Joint Committee on Cancer guidelines. The clinicopathological findings of the patients are presented in Table 1. All of the patients were women, and the median age was 46 years (range, 25 to 83 years). The mean number of metastatic lymph nodes was 4.95 (range, 1 to 27; median, 3.00). The survival data were obtained from a review of the patient's chart in our hospital and from the National Cancer Registry. The mean follow-up time was 45.5 (range, 1 to 63) months. Recurrence-free survival was defined as the number of months from diagnosis to the occurrence of an event (local recurrence/metastasis). Plasma specimens were also obtained from 189 healthy individuals (109 men and 80 women) as controls.
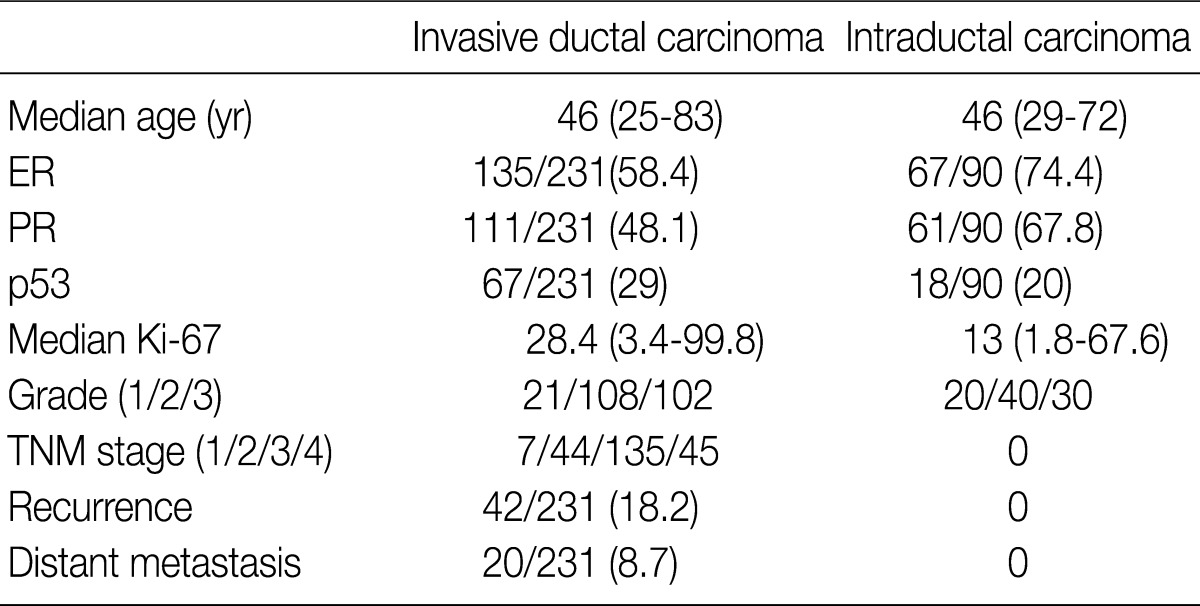
Clinicopathological parameters of the intraductal and invasive breast carcinomas
For DNA extraction, we used fresh-frozen tissues for invasive carcinoma and formalin-fixed, paraffin-embedded tissues for intraductal carcinoma. The H&E-stained sections were reviewed to confirm the tumor areas and tumor volume before DNA extraction. The tumor-rich areas (>75%) were extracted from 0.1% methylene blue-stained serial 10 µm tissue sections, and the areas corresponding to tumor parenchyma were carefully microdissected from the surrounding stromal tissues. DNA was extracted using a DNeasy tissue kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions.
Assembly of tissue microarrays
The specimens were fixed in 10% buffered formalin, and embedded in paraffin. The representative areas on the H&E-stained sections were carefully selected and marked on the individual paraffin blocks. Two tissue cores (1 mm in diameter) were obtained from the regions of interest in each tumor block. The tissue cores were then inserted in a recipient paraffin block according to the manufacturer's instructions (Isu Abxis Co., Ltd., Cambridge, UK).
Immunohistochemistry
After deparaffinization and rehydration, the 4 µm thick sections on the silane-coated slides were heat-pretreated in a citrate buffer (pH 7.3 at 92℃ in a microwave oven) and immunostained using specific antibodies directed against p16 (G175-405, Pharmingen, Hamburg, Germany), Her-2 (CB11, Novocastra Laboratories, Newcastle, UK), estrogen receptor (ER; 6F11, Novocastra Laboratories), progesterone receptor (PR; 1A6, Novocastra Laboratories), p53 (Zymed, South San Francisco, CA, USA), and Ki-67 (MIB I, Dianova, Hamburg, Germany). An avidin-biotin technique was applied using 3,3'-diaminobenzidine (DAB) for visualization and hematoxylin as the nuclear counterstain. Visual estimation of the nuclear and/or cytoplasmic staining of p16 was performed. A cell was considered positive for p16 protein if it contained staining within the nucleus, cytoplasm, or both the nucleus and cytoplasm. The number of stained tumor cells was determined semiquantitatively. The proportion of positive cells was classified into five categories: 0 (1-4% positive tumor cells), 1 (5-10%), 2 (10-50%), 3 (51-80%), or 4 (>80%). The cases with more than 5% positive cells were defined as positive. The staining intensity was scored as follows: 1 (weak), 2 (moderate), or 3 (strong). The scores for both parameters were multiplied to generate the immunoreactivity score (IS) for each case. As a result, the IS range was from 0 to 12. Each lesion was examined and scored separately by two pathologists, and the cases with discrepant scores were discussed until agreement was reached between the pathologists.
Protein expression was determined in a blinded manner for the methylation analyses and vice versa. The distribution of Her-2 was assessed according to the following scoring system: 0, no immunoreactivity or immunoreactivity in less than 10% of the tumor cells; 1+, faint and incomplete staining of more than 10% of the tumor cells; 2+, weak to moderate complete membrane immunoreactivity in more than 10% of the tumor cells; 3+, moderate to strong complete membrane immunoreactivity in more than 30% of the tumor cells. The semiquantitative histochemical scores were used to record the results of the ER and PR staining according to the Allred Scoring System.18 The p53 staining in the cell nuclei was assessed, and the patients with staining in more than 5% of the tumor cells were regarded as positive.
DNA modification and methylation analysis
One microgram of genomic DNA was modified using sodium bisulfite with the EZ DNA methylation kit (Zymo Research, Orange, CA, USA) according to the manufacturer's instructions. Modified DNA was stored in aliquots at -20℃ until required. The methylation status of the p16INK4a gene was determined by methylation-specific PCR (MSP) using two pairs of primers, as described by Herman et al.3 The MSP primers specific for methylated p16INK4a gene were 5'-TTATTAGAGGGTGGGGCGGATCGC-3' (sense) and 5'-GACCCCGAACCGCGACCGTAA-3' (antisense), and the primers used for the unmethylated p16INK4a gene were 5'-TTATTAGAGGGTGGGGTGGATTGT-3' (sense) and 5'-CCACCTAAATCAACCTCCAACCA-3' (antisense). Amplification was carried out over 35 cycles (1 minute at 94℃, 1 minute at the annealing temperature, and 1 minute at 72℃), followed by 4 minutes at 72℃. The PCR product sizes were 150 bp and 234 bp for the methylated and unmethylated segments, respectively. CpGenome Universal Unmethylated DNA (S7822, Millipore, Billerica, MA, USA) was used as a positive control for the unmethylated alleles, and CpGenome Universal Methylated DNA (S7821, Millipore) was used as a positive control for the methylated alleles. The PCR products were visualized on 2% agarose gels stained with ethidium bromide.
Quantitative real-time MSP of plasma
Quantitative real-time MSP was performed. The primer sequences used to amplify the actin gene were 5'-TGGTGATGGAGGAGGTTTAGTAAGT-3' (sense) and 5'-AACCAATAAAACCTACTCCTCCCTTAA-3' (antisense) were used in conjunction with a fluorogenic probe 5'-(FAM)-ACCACCACCCAACACACAATAACAAACACA-(TAMRA)-3', and the primer sequences used for the methylated p16INK4a gene were 5'-GGGGAGAGTAGATAGCGGGC-3' (sense) and 5'-AACCAATCAACCGAAAATTCCATA-3' (antisense) in conjunction with a fluorogenic probe 5'-(FAM)-TACTCCCCGCCGCCGACTCCAT-(TAMRA)-3'. Each 10 µL PCR reaction volume contained 5 µL TaqMan Universal PCR Master Mix No AmpErase UNG (Applied Biosystems, Foster City, CA, USA), 1 pM of each primer, a 0.025 pM probe, and 2 µL template DNA. Assays were run on an ABI Prism 7900 Sequence Detection System (Applied Biosystems). The PCR conditions were as follows: 2 minutes at 50℃, 10 minutes at 95℃, followed by 50 cycles of 15 seconds at 95℃, and 1 minute at 60℃. Real-time quantitative PCR reactions were performed for the detection and quantitation of the bisulfite unconverted methylated version of the p16INK4a gene and the bisulfite-converted unmethylated version of the actin gene. Serial dilutions of methylated or unmethylated control genomic DNAs (Millipore) were used to construct standard curves. The methylation index (MI) in each sample was calculated using the following equation:19
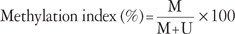
MI values greater than 0.1% were considered positive.
Statistical analysis
Statistical analyses of the immunohistochemical abnormalities were performed using the two-tailed Fisher's exact test with a p-value set as <0.05 to indicate significance. Fisher's exact tests were used to compare aberrant p16 gene expression and p16INK4a methylation with the clinicopathologic parameters of breast cancer. Comparison of the amount of methylated p16INK4a DNA in the plasma between the invasive breast cancer patients and normal controls was performed using the Student's t-test (two-tailed). All statistical comparisons were performed using the statistical software, SPSS (SPSS Inc., Chicago, IL, USA).
RESULTS
Methylation analysis
We analyzed the pattern of p16INK4a methylation in the 231 invasive breast carcinomas and 90 intraductal carcinomas. The results of these experiments are summarized in Table 2 and representatively illustrated in Fig. 1. The p16INK4a promoter was frequently hypermethylated, not only in the invasive tumor samples but also in the intraductal carcinoma specimens. In many of the specimens examined (122/231 [52.8%] for invasive ductal carcinoma and 52/90 [57.8%] for intraductal carcinoma) we observed the presence of the amplification band corresponding to the methylated target sequence. We also detected the unmethylated allele in 114 invasive cancer and 53 intraductal carcinoma specimens, suggesting contamination of the dissected tumor samples with normal cells. Ninety-eight cases of invasive cancer and 43 cases of intraductal carcinoma were fully unmethylated.
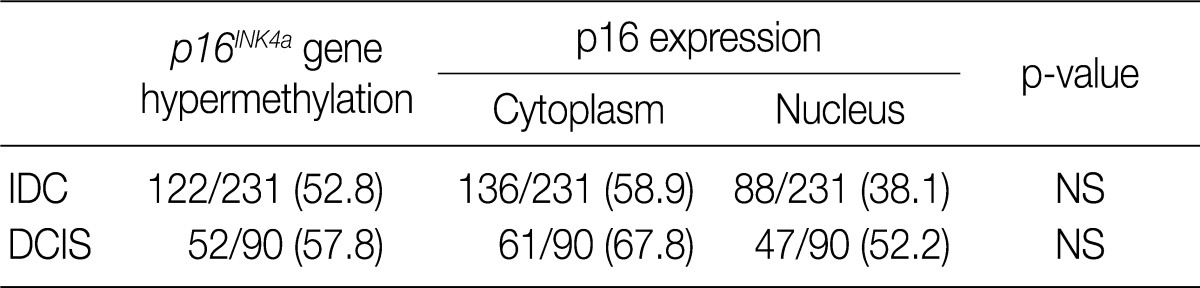
Frequency of p16INK4a hypermethylation and p16 expression in the intraductal and invasive breast carcinomas
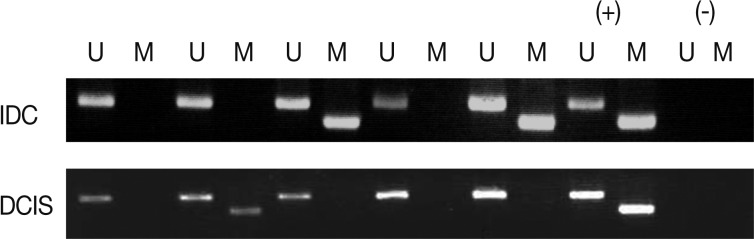
Methylation-specific polymerase chain reaction for the p16INK4a gene in the invasive and intraductal carcinomas with positive and negative controls. IDC, invasive ductal carcinoma; DCIS, ductal carcinoma in situ; U, unmethylated; M, methylated; (+), positive control; (-), distilled water.
Immunohistochemical analysis
The majority of breast carcinoma samples moderately or strongly expressed p16 in either the infiltrating or intraductal component, or both (Table 2, Fig. 2). As shown in Table 2, immunohistochemical analysis showed that 145 of 231 invasive carcinomas (62.8%) and 63 of 90 intraductal carcinomas (70%) overexpressed the p16 protein. The reactivity was predominantly nuclear and cytoplasmic or cytoplasmic alone (Fig. 2). The overexpression of p16 in the invasive carcinomas was significantly associated with a high histologic grade, negative ER and PR status, p53 immunoreactivity, and high Ki-67 proliferation index (Table 3). The IS score of the p16 immunoreactivity also correlated with the above variables. No correlation of p16 expression with clinical stage, HER2/neu status, or disease free survival was found.
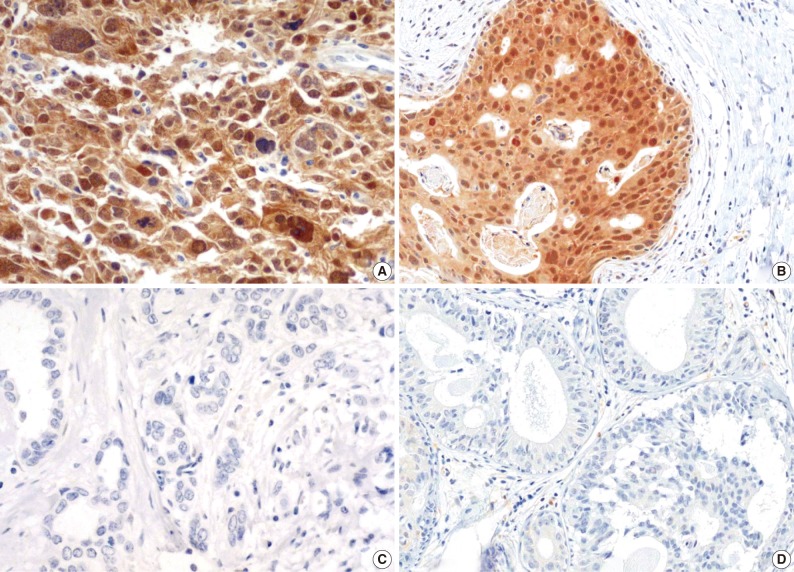
Immunohistochemical staining for p16 frequently reveals high expression in the invasive (A) and intraductal (B) carcinomas of the breast. The photomicrographs reveal moderate or strong expression of p16 in either the nucleus and cytoplasm or the cytoplasm alone. Negative expression in the invasive (C) and intraductal (D) carcinomas of the breast.
Correlation between the clinicopathological parameters and p16 expression of the intraductal and invasive breast carcinomas
Relationship between MSP and immunohistochemical analyses
In our study, there was no clear association between promoter hypermethylation of p16INK4a and p16 expression. In the invasive carcinomas, 78 of 122 tumors with methylation revealed p16 overexpression (p=0.7) while 44 tumors showed loss of protein. In the intraductal carcinomas, 34 of 52 tumors with methylation had high expression of p16 (p=0.269), while 18 tumors showed loss of protein (Table 2).
Plasma concentration of methylated p16INK4a promoter DNA in the breast carcinoma patients
Peripheral blood plasma was obtained from 200 invasive breast cancer patients before surgery and from 189 healthy donors, which were used as normal controls. The amount of methylated p16INK4a gene copies was examined using the quantitative real-time MSP approach and was expressed as the MI (%). Methylated p16INK4a was found in the plasma of 69 of the 200 (34.5%) invasive breast cancer patients and in 2 of the 189 (1%) normal controls. Among the control group, all of the subjects with methylated p16INK4a were men. The mean MI for p16INK4a was 6.37% (range, 0 to 85%) in the cancer patients and 0.06% (range, 0 to 7.3%) in the normal controls (Fig. 3). The mean concentration of methylated p16INK4a in the plasma of the invasive breast cancer patients was higher than that in the normal healthy control group, which was statistically significant (t-test, p=0.016).
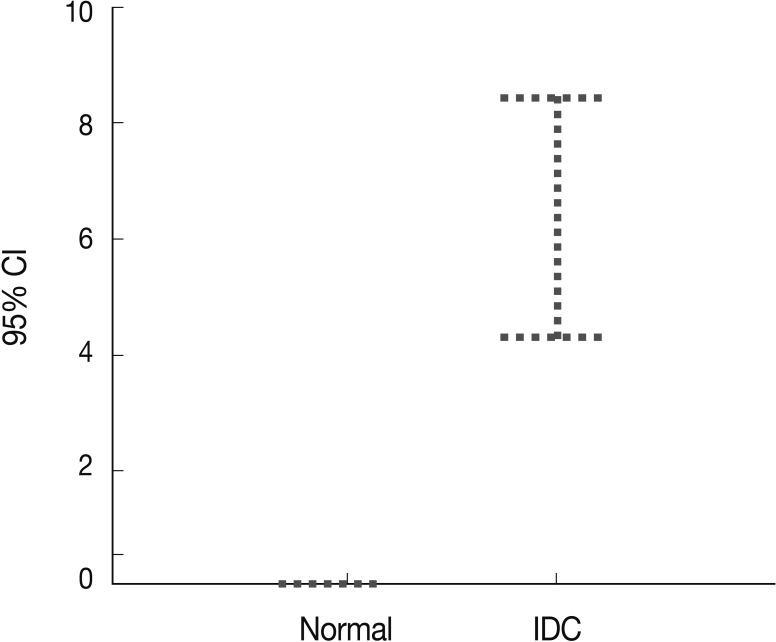
p16INK4a gene hypermethylation in the plasma of the breast cancer patients (n=200) and normal healthy controls (n=189) is determined using quantitative real-time polymerase chain reaction. p16INK4a methylation is significantly higher in the plasma of the cancer patients than the normal controls. CI, confidence interval; IDC, invasive ductal carcinoma.
DISCUSSION
p16INK4a promoter methylation is variably present in breast carcinoma with a prevalence ranging from 4% to 68.4%,3,20-22 and our present study revealed a relatively high frequency of methylation of 52.8% (122/231) and 57.8% (52/90) in the invasive and intraductal tumors, respectively. The high prevalence of p16INK4a methylation in both the invasive and intraductal carcinomas may indicate that the epigenetic alterations of the p16INK4a gene play a significant role in the early stage of mammary carcinogenesis. Recently, Liu et al.23 suggested that p16INK4a methylation in various intraductal proliferative lesions, such as usual ductal hyperplasia, flat epithelial atypia, atypical ductal hyperplasia, low grade intraductal carcinomas, and high grade intraductal carcinomas, was related to the progression of intraductal epithelial proliferative lesions of the breast.
The p16INK4a gene is epigenetically silenced in many human tumors; however, regardless of the cut-off point of immunoreactivity, our study did not show a firm association between methylation and protein expression. Similar findings have been reported in breast carcinomas and in other malignancies.4,24-26 There are several possible explanations for this discrepancy between immunohistochemistry and promoter hypermethylation. For example, the discrepancy may be caused by tumor heterogeneity. It is possible that both p16-positive cells and -negative cells were present in some cases, and that the predominant p16-positive cells were analyzed for p16INK4a promoter hypermethylation. Another explanation may be the regulation of protein expression by post-transcriptional alterations other than promoter methylation. Moreover, the immunoreactivity of p16 proteins has been reported to be variable according to different anti-p16 antibodies. The G175-405 antibody used in our study has been reported to be the most specific.27
The present study showed that p16 protein overexpression was correlated with several phenotypic parameters of the tumors indicative of a poor prognosis, although it was not associated with disease-free survival or overall survival. This suggested that the high expression of p16 correlated with an aggressive tumor phenotype. Some reports in the literature are in agreement with the above findings,5-7,28 whereas other reports did not find any correlation.29 The pathophysiological role of p16 in the oncogenesis of breast carcinoma should be focused in further investigation. p16 is a tumor suppressor, and it is accepted that the loss of p16 expression is related to tumorigenesis. Therefore, our data could be interpreted as overexpression of p16 as an attempt to compensate for the oncogenic changes of other proteins, such as retinoblastoma or TP53.30 In the present study, high expression of p16 correlated with both p53 protein positivity and a high Ki-67 proliferation index. The p16 overexpression in the patients with high proliferative activity may indicate that p16 is inactive or not sufficient to limit cell growth in breast carcinomas.
By using a quantitative PCR approach, we found that methylated p16INK4a DNA was present in the plasma of both the cancer patients and healthy subjects. However, the invasive breast cancer patients had significantly higher frequencies and concentrations of methylated p16INK4a DNA than the normal controls. Our results suggest that a quantitative measurement of methylated p16INK4a DNA concentrations in the plasma of cancer patients might be a useful tool for cancer screening and monitoring of treatment effectiveness. Nevertheless, a large-scale prospective study of plasma from cancer patients is necessary to further investigate the feasibility of methylated p16INK4a DNA as a tumor screening marker in invasive breast cancer.
In conclusion, the high frequency of p16INK4a methylation in invasive breast carcinoma and intraductal carcinoma suggest that p16INK4a methylation could be a common and early event in breast carcinogenesis. The overexpression of p16 in the invasive carcinomas was correlated with several phenotypic parameters of the tumors indicative of poor prognosis, although it was not associated with disease free survival or overall survival. Therefore, this suggested that the high level of p16 expression may be an indicator of poor prognosis. Moreover, the concentrations of p16INK4a methylated DNA was significantly higher in the plasma of the invasive breast cancer patients compared with the healthy individuals, suggesting that quantitative measurement of plasma methylated p16INK4a gene concentrations might be a useful and non-invasive tool for cancer screening and monitoring treatment response.
Notes
No potential conflict of interest relevant to this article was reported.