KRAS Mutation Detection in Non-small Cell Lung Cancer Using a Peptide Nucleic Acid-Mediated Polymerase Chain Reaction Clamping Method and Comparative Validation with Next-Generation Sequencing
Article information
Abstract
Background
KRAS is one of commonly mutated genetic "drivers" in non-small cell lung cancers (NSCLCs). Recent studies indicate that patients with KRAS-mutated tumors do not benefit from adjuvant chemotherapy, so there is now a focus on targeting KRAS-mutated NSCLCs. A feasible mutation detection method is required in order to accurately test for KRAS status.
Methods
We compared direct Sanger sequencing and the peptide nucleic acid (PNA)-mediated polymerase chain reaction (PCR) clamping method in 134 NSCLCs and explored associations with clinicopathological factors. Next-generation sequencing (NGS) was used to validate the results of discordant cases. To increase the resolution of low-level somatic mutant molecules, PNA-mediated PCR clamping was used for mutant enrichment prior to NGS.
Results
Twenty-one (15.7%) cases were found to have the KRAS mutations using direct sequencing, with two additional cases by the PNA-mediated PCR clamping method. The frequencies of KRAS mutant alleles were 2% and 4%, respectively, using conventional NGS, increasing up to 90% and 89%, using mutant-enriched NGS. The KRAS mutation occurs more frequently in the tumors of smokers (p=.012) and in stage IV tumors (p=.032).
Conclusions
Direct sequencing can accurately detect mutations, but, it is not always possible to obtain a tumor sample with sufficient volume. The PNA-mediated PCR clamping can rapidly provide results with sufficient sensitivity.
KRAS mutation is common in non-small cell lung cancer (NSCLC), occurring in 20% to 25% of cases.1 Approximately 97% of KRAS mutations in NSCLCs involve codon 12 or 13.2 KRAS mutations are associated with resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), gefitinib, and erlotinib.3,4 Although further studies are required, the multi-kinase inhibitor, sorafenib, may be a potential treatment for KRAS-mutated lung cancer.5 Recently, the molecular testing in NSCLCs has become an important part of treatment planning. EGFR mutation status is a powerful predictive marker for response to EGFR TKIs.6,7 The College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology recommend EGFR testing for adenocarcinomas. Because KRAS and EGFR mutations are mutually exclusive, a KRAS assay may be performed initially to exclude KRAS-mutated tumors from EGFR mutation testing.8 While EGFR mutation occurs within exons 18 to 21, KRAS mutations only occur within three codons; thus, KRAS mutation testing can be performed more efficiently than EGFR mutation testing.
A number of tests are available for the detection of KRAS mutations. Conventional polymerase chain reaction (PCR) amplification followed by direct Sanger sequencing has been widely used, and direct sequencing can detect any type of mutation within exon 2. However, direct sequencing is relatively less sensitive when the percentage of tumor cells in the sample is low.
The peptide nucleic acid (PNA)-mediated PCR clamping method has recently been developed to detect KRAS mutations.9 PNA is a synthetic DNA analog in which the phosphoribose backbone has been replaced by a peptide-like repeat of the (2-aminoethyl)-glycine chain. PNA/DNA hybrids have a higher thermal stability than corresponding DNA/DNA hybrids.10 Therefore, PNA that is complementary to a wild-type sequence can inhibit primer annealing and prevent amplification of the wild-type gene. The PNA-mediated PCR clamping method is more sensitive than direct sequencing, and has the ability to detect mutations in samples comprising less than 1% mutant alleles.11,12,13,14,15
The aim of this study was to compare the two methods of KRAS mutation detection using 134 NSCLC samples. Next-generation sequencing (NGS) was used to validate the results.
MATERIALS AND METHODS
Patients
One hundred and thirty-four cases of primary NSCLC were obtained from pathology files spanning from 2012 to 2013, including seventy-six biopsy samples of lung or lymph nodes, fifty-seven samples from surgical resection of lung or lymph nodes, and one pleural fluid sample. Clinical information including cancer stage and smoking history were obtained based on clinical records. This study was approved by the Institutional Review Board at the Samsung Medical Center (Seoul, Korea).
DNA extraction
Genomic DNA was extracted from 5-µm-thick sections of 10% neutral formalin-fixed, paraffin-embedded tumor tissue blocks using the QIAamp DNA mini kit (Qiagen, Hilden, Germany). The concentration and purity of the extracted DNA was determined by a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). The extracted DNA was stocked at -20℃ until use.
Genomic DNA from the A549 cell lines was diluted with the DNA from the HeLa cells (New England Biolabs, Hitchin, UK) to give mutant/wild-type ratios of 0%, 0.01%, 0.05%, 0.1%, 0.5%, 1%, 10%, and 100%. The manufactured DNA targets were stored at -20℃ until use.
Direct Sanger sequencing of KRAS
Mutational analysis of KRAS exon 2 was performed by direct Sanger sequencing of PCR products amplified from genomic DNA. PCR was performed in a 20-µL volume containing 100 ng of template DNA, 10× PCR buffer, 0.25 mM dNTPs, 10 pmol primers, and 1.25 U Taq DNA polymerase (iNtRON, Daejeon, Korea). PCR products were electrophoresed on 2% agarose gels and purified with a QIAquick PCR purification kit (Qiagen). Bidirectional sequencing was performed using the BigDye Terminator v1.1 kit (Applied Biosystems, Foster City, CA, USA) on an ABI 3130xl genetic analyzer (Applied Biosystems). Chromatograms were manually reviewed for sequence analysis. Confirmatory re-sequencing from replicate PCR amplification reactions was performed for any sequences that were ambiguous. The results were marked as mutation positive if a mutation was detected in both the forward and reverse DNA strand.
PNA-mediated clamping PCR of KRAS
Assays for the detection of seven different KRAS variants were obtained through the PNAClamp KRAS, Mutation Detection kit (Panagene Inc., Daejeon, Korea). All reactions were performed in a 20-µL volume using 10 ng template DNA, a primer and PNA probe set, and SYBR Green PCR master mix. All reagents needed were included with the kit. The real-time PCR reaction of PNA-mediated clamping was performed using a CFX 96 (Bio-Rad, Hercules, CA, USA). PCR cycling conditions were a 5-minute hold at 94℃ followed by 40 cycles of 94℃ for 30 seconds, 70℃ for 20 seconds, 63℃ for 30 seconds, and 72℃ for 30 seconds. Detection of each of seven mutations in the KRAS gene was possible using one-step PNA-mediated real-time PCR clamping. In this assay, PNA probes and DNA primers were used together in the clamping reaction. Positive signals were detected by intercalation of SYBR Green fluorescent dye. The PNA probe sequence, which was complementary to wild-type DNA, suppressed amplification of the wild-type target, thereby enhancing preferential amplification of mutant sequences by competitively inhibiting DNA primer binding to wild-type DNA. PCR efficiency was determined by measuring the threshold cycle (Ct) value. Ct values for the control and mutation assays were obtained by observing the SYBR Green amplification plots. Mutation status was determined based on a Ct value difference greater than 2 between the control and sample.
Next-generation sequencing
PCR amplification for conventional NGS
Target samples were analyzed by next-generation sequencing for KRAS mutations with the Cancer Panel on a GS Junior Sequencer (Roche Diagnostics, Mannheim, Germany). For conventional 454 targeted resequencing, 30 ng of genomic DNA was used for PCR of the KRAS panel (SeaSun Biomaterials, Daejeon, Korea). Subsequent processing of the samples was performed according to the manufacturer's protocol.
Enrichment PCR for mutant-enriched NGS and sequencing library preparation
To increase the resolution of low level somatic mutant molecules within a high background of wild-type molecules, Insight Onco Panel for KRAS (SeaSun Biomaterials) was used for mutant enrichment PCR according to the manufacturer's instructions. The mutant-specific enrichment PCR was performed using 30 ng of genomic DNA and subsequent processing of the samples was performed according to the manufacturer's protocol. Nonspecific PCR products were removed by Agencourt AMPure XP beads (Beckman Coulter, Vienna, Austria) using a 1:1 DNA to bead ratio.
Sequencing library preparation PCR was performed using 2 µL of purified PCR products from enrichment PCR amplification as a template, KRAS codon 12/13 Insight 2× Seq Lib Pep Premix (SeaSun Biomaterials) and each barcoded primer pair. The sequencing adaptor with multiplex identifier (MID) was conjugated using the manufacturer's protocol. Unwanted short fragments were removed by Agencourt AMPure XP beads (Beckman Coulter) using a 1:1 DNA to bead ratio.
Quantification and normalization of sequencing amplicons
Purified amplicons were quantitated by Pico-Green (Life Technologies, Carlsbad, CA, USA) utilizing an external Infinite F200Pro fluorometer (Tecan, Grodig, Austria) with Magellan v7.0 Software (Tecan). Based on the standard concentrations, the signals were directly translated to ng/µL and the coefficient of determination (validation criteria r2>0.99) was calculated from eight DNA standards in a range from 0 to 100 ng/µL. For emulsion PCR amplification, the concentrations from the amplicons were converted in molecules/µL using the associated amplicon length. The manufactured DNA pools were stored at -20℃ until further use.
Ultradeep pyrosequencing
Pyrosequencing was carried out according to the manufacturer's protocol for amplicons using the GS Junior System (Roche Diagnostics). Emulsion PCR, breaking, and bead enrichment was carried out using the GS Junior Titanium emPCR Kit Lib-L, emPCR Reagents Lib-L kit, Oil and Breaking kit, and the Bead Recovery Reagents kit according to the supplier's instructions (Roche Diagnostics). For emulsion PCR, we used a copy-per-bead ratio of 0.5. Enrichment of DNA-carrying beads was done with magnetic beads and a magnetic particle collector (Invitrogen, Carlsbad, CA, USA). To evaluate the amount of enriched beads, counting was performed using the GS Junior Bead Counter (Roche Diagnostics). Finally, we loaded 100,000-500,000 beads onto the PicoTiterPlate (Roche Diagnostics). Sequencing was carried out according to standard Roche/454 protocols using the GS Titanium Sequencing kit (Roche Diagnostics) and the GS Junior device.
Data analysis
Processed and quality-filtered reads were analyzed with the GS Amplicon Variant Analyzer. Sequencing reads data were visualized using the GS Amplicon Variant Analyzer (Roche Diagnostics). The KRAS amplicons (excluding adaptors and MIDs) were used as the references to align amplicon reads, template-specific portions of the fusion primers were considered primer A and primer B, and the known mutations of the samples selected were defined as substitutions relative to the reference sequence. Correspondence of samples and MID tags was specified and, as the MID was present in primer A, a "Primer 1 MID" encoding multiplexer was used to demultiplex the reads.
Statistical analysis
Statistical differences between the two methods were analyzed by the McNemar test. The chi-square test or Fisher's exact test was used to compare the qualitative data. The t-test was used to compare means. Statistical calculation was performed with SPSS ver. 18 (SPSS Inc., Chicago, IL, USA). A p<.05 was considered statistically significant.
RESULTS
The mean age of patients was 63 years. Seventy-eight of the patients were male and fifty-six were female. The majority of cases were adenocarcinomas (n=124), though there were also four cases of mucinous adenocarcinoma, three cases of squamous cell carcinoma, and one case of pleomorphic carcinoma. Thirty-four cases had stage I tumors, fifteen cases had stage II tumors, nineteen cases had stage III tumors, and sixty-six cases had stage IV tumors.
Twenty-one (15.7%) of the 134 cases were found to have a KRAS missense mutation using direct Sanger sequencing. However, when the samples were tested using the PNA-mediated PCR clamping method, two additional cases (case nos. 92 and 133) were found to have a KRAS mutation. Chromatograms of discordant cases were reviewed, and clearly showed no abnormal peaks (Fig. 1). However, the difference between the two methods was not statistically significant (p=.5). All missense mutations were located on codon 12, and 18 mutations were located on the 35th base (Fig. 2A). This frequency was similar to that reported in the Catalogue of Somatic Mutations in Cancers (COSMIC) (http://www.sanger.ac.uk/cosmic) (Fig. 2B).
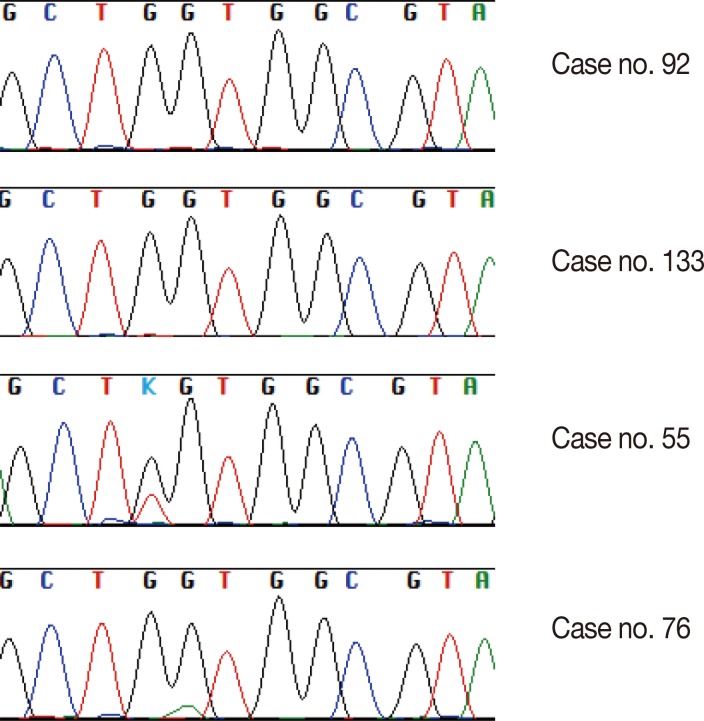
Part of a direct sequencing chromatogram showing codon 12 to codon 14. No secondary peaks are identified in case no. 92 or case no. 133 compared with case no. 55 and case no. 76, which have missense mutations in codon 12.
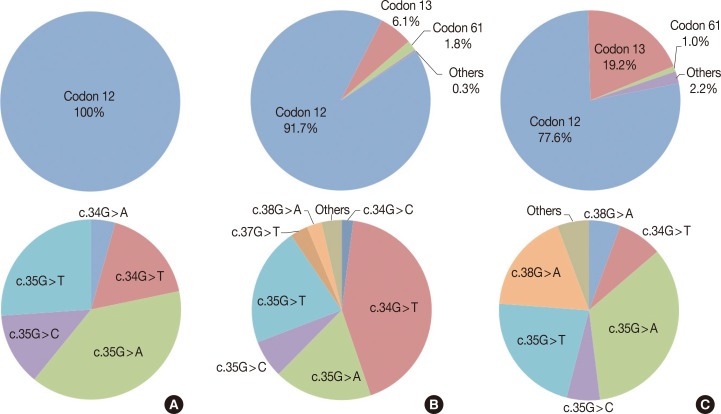
(A) Proportion of KRAS mutation genotypes in this study (n=23). All mutatons are located in codon 12. (B) COSMIC data on the proportion of lung cancer KRAS mutations (n=3,743). Proportion of each genotype in this study is similar to COSMIC data. (C) COSMIC data on the proportion of large intestine cancer KRAS mutation (n=17,853). Proportion of genotype is also similar between lung cancer and large intestine cancer.
Case no. 92 was diagnosed as adenocarcinoma, with a tumor size of 1 cm and a tumor volume of approximately 20% (Fig. 3A). The sample of case no. 133 was taken from an adenocarcinoma that had been resected following neoadjuvant chemotherapy. This tumor showed a diffuse desmoplastic reaction with scattered tumor cells, with tumor comprising only about 5% of the resected tissue (Fig. 3B).
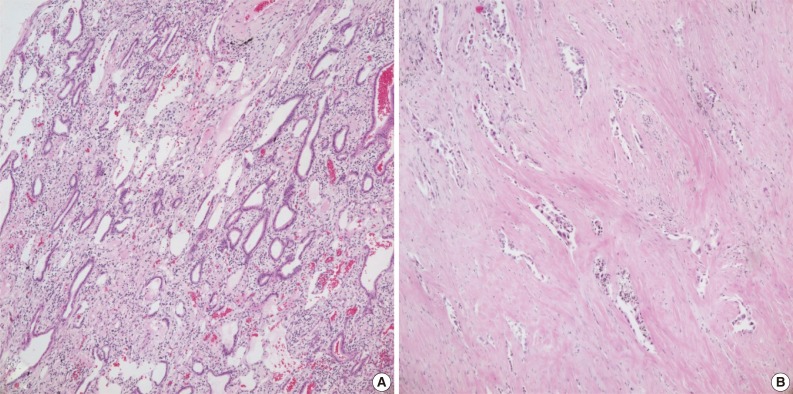
Hematoxylin and eosin staining. (A) Case no. 92. Heavy inflammatory infiltrates are seen around the tumor glands. DNA of inflammatory cells can lower the proportion of tumor DNA. (B) Case no. 133. Tumor cells are sparsely distributed within the fibrous stroma. Tumor volume is reduced after neoadjuvant chemoradiotherapy.
We validated the discrepant cases by NGS with and without mutant enrichment using PNA-clamping. We compared the sensitivity of conventional NGS and mutant-enriched NGS by diluting control DNA. KRAS mutations could be detected by conventional NGS when the mutant allele comprised more than 1% of the sample. However, following enrichment, the mutation could be detected when the mutant allele comprised only 0.05% of the sample (Fig. 4).
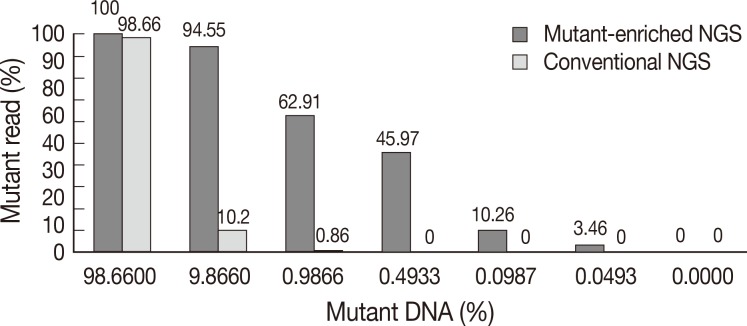
Comparison of conventional next-generation sequencing (NGS) and mutant-enriched NGS. While more than 10% of mutant DNA is needed for conventional NGS to detect mutations, mutant-enriched NGS can detect as low as 0.05% of mutant DNA.
Using conventional NGS, the percentage of KRAS mutations detected in case no. 92 and case no. 133 was 2% and 4%, respectively. The mutant KRAS allele was increased in mutant-enriched NGS up to 90% and 89% in each case, respectively (Table 1). NGS was also performed on a wild-type case (case 2) for comparison.
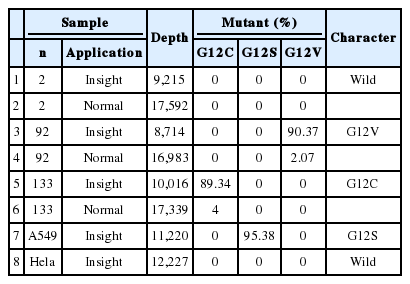
Next-generation sequencing results
Clinicopathologic characteristics of 23 cases are summarized in Table 2. We analyzed the correlation between clinical data and KRAS mutation status (Table 3). KRAS mutations tended to be present in the tumors of smokers (p=.011). Of the 23 patients who had a KRAS mutated tumor, five (9.0%) were women and 18 (23%) were men. Cancer stage was also found to be related to KRAS mutation; stage IV tumors were more likely to have a KRAS mutation (p=.032).

Clinicopathologic data of KRAS-mutated adenocarcinoma cases
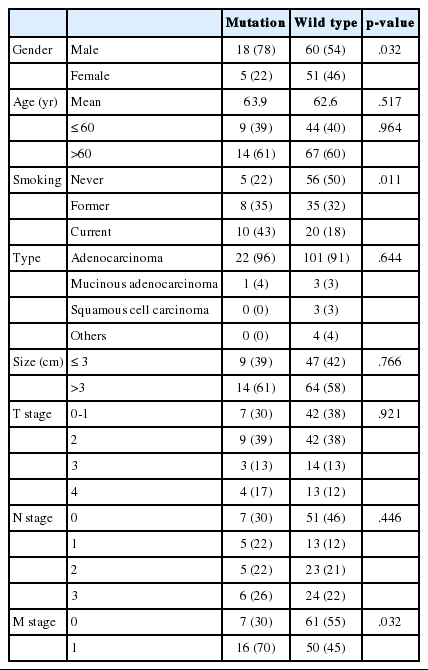
Statistical differences between KRAS-mutated and wild-type cases
DISCUSSION
Of 134 cases of NSCLC, KRAS mutations were detected in 21 cases using direct sequencing and in 23 cases using a PNA-mediated PCR clamping method. Although the difference was not statistically significant, two additional cases of KRAS mutation were detected with the PNA-mediated PCR clamping method. The mutation status of the two discordant samples was confirmed using NGS. The percentage of mutant alleles in both samples was less than 5%. With such a low percentage, a mutation peak would be indistinguishable from background noise by direct sequencing.11,12 One of these cases had a low viable tumor volume due to neoadjuvant chemotherapy. The tumor volume of the other case was not as low. Several factors may have contributed to the lower mutant proportion. DNA of inflammatory cells around the tumor can dilute tumor DNA. Tumor volume may have been diminished on serial sections of paraffin block. In addition, tumor heterogeneity may be present. In conventional NGS, the percentage of mutations detected was approximately the same as the percentage of mutant alleles in the sample. This allows one to reliably determine tumor heterogeneity. A proportion of mutant alleles in a sample of less than 1% means that the mutation cannot be detected using conventional NGS. In contrast, as few as 0.05% mutant alleles can be detected after enrichment for the mutant alleles using the PNA clamping method.
Similar to previous studies,1,16 our results show an association between KRAS mutation and smoking status. KRAS mutations were more frequent in male patients than female patients. It is possible that there is a sex bias in this relationship, as 71 of 73 smokers in the sample were men. Tumor stage was also related to KRAS mutation according to our data. However, in other reports, stage was not significantly correlated with KRAS mutation.17,18,19,20,21,22 In one meta-analysis,23 KRAS mutation was found to be a statistically significant prognostic factor in lung adenocarcinoma. There have been multiple reports recently addressing the relationship between KRAS mutation status and survival, with various outcomes (Table 4). Since the mutant-enriched sequencing method is more sensitive than direct sequencing, the KRAS mutation rate is relatively higher in reports using mutant-enriched sequencing. Racial difference and proportion of adenocarcinoma included in study also affected KRAS mutation rate. Studies of a Caucasian population or adenocarcinoma have higher KRAS mutation rates than studies of Asian populations or any histologic type of NSCLC.
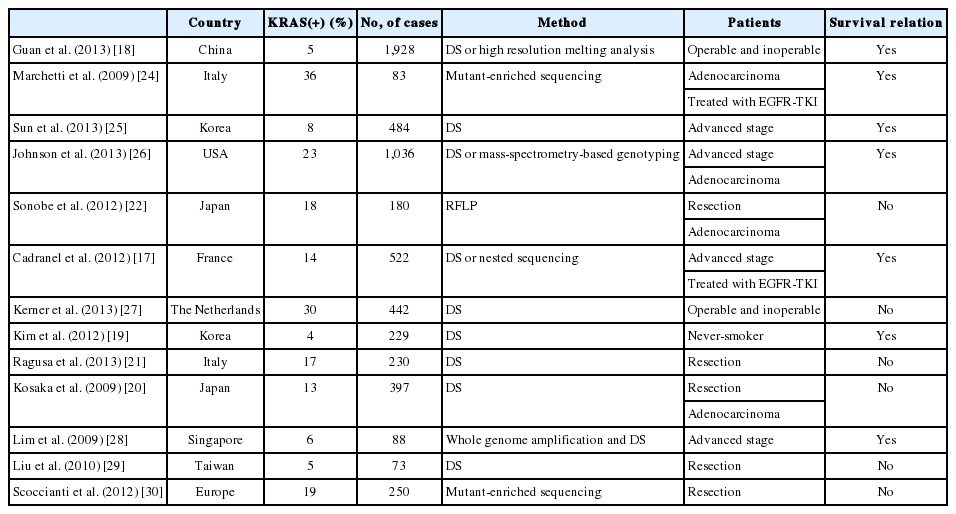
Recent studies examining the relationship between survival outcomes and KRAS mutation in non-small cell lung cancer
In this study, the number of cases was too small to establish statistical significance between the two methods. Survival analysis could not be performed due to the short follow-up period. Further large-scale studies may be required to assess the differences between the two methods of KRAS mutation detection in NSCLC.
As targeted therapy for KRAS mutation continues to develop, testing in NSCLCs will become more important. Direct sequencing can accurately detect mutations when the percentage of tumor cells in the analytical sample is sufficiently high. However, it is not always possible to obtain samples with enough volume to undertake these tests. In these situations, methods other than direct sequencing are required. The PNA-mediated PCR clamping method can quickly provide results and is sufficiently sensitive in this situation.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Science, ICT & Future Palnning (MSIP) (NRF-2013R1A2A2A01068922).
Notes
No potential conflict of interest relevant to this article was reported.