Extrapulmonary Lymphangioleiomyoma: Clinicopathological Analysis of 4 Cases
Article information
Abstract
Background
Lymphangioleiomyomatosis (LAM) is a slowly progressive neoplastic disease that predominantly affects females. Usually, LAM affects the lung; it can also affect extrapulmonary sites, such as the mediastinum, the retroperitoneum, or the lymph nodes, although these locations are rare. A localized form of LAM can manifest as extrapulmonary lesions; this form is referred to as extrapulmonary lymphangioleiomyoma (E-LAM). Due to the rare occurrence of E-LAM and its variable, atypical location, E-LAM is often difficult to diagnose. Herein, we report the clinicopathological information from four E-LAM cases, and also review previous articles investigating this disease.
Methods
Four patients with E-LAM were identified at the Samsung Medical Center (Seoul, Korea) from 1995 to 2012. All E-LAM lesions underwent surgical excision.
Results
All patients were females within the age range of 43 to 47 years. Two patients had para-aortic retroperitoneal masses, while the other two patients had pelvic lesions; two out of the four patients also had accompanying pulmonary LAM. In addition, no patient displayed any evidence of tuberous sclerosis. Histologically, two patients exhibited nuclear atypism with cytologic degeneration.
Conclusions
E-LAM should be considered in the differential diagnosis of patients presenting with pelvic or para-aortic masses. We also conclude that further clinical and pathological evaluation is needed in patients with E-LAM and nuclear atypism.
Lymphangioleiomyomatosis (LAM) is a neoplastic disease with slowly progressive behavior1 that generally arises in the lung and predominantly affects females. Moreover, tuberous sclerosis (TS) is known to be associated with LAM. TS is an autosomal dominant syndrome characterized by benign brain neoplasms or hamartomas, retinal glial hamartomas, cardiac rhabdomyomas, pulmonary LAM (P-LAM), and renal angiomyolipomas. A sporadic form of LAM, defined as LAM with no relation to TS, has been detected in up to three to five women per million in the general population.2 Usually, LAM is detected in the lungs and is accompanied by symptoms such as a persistent cough, hemoptysis, and chyloptysis. Unfortunately, no effective treatments for LAM have yet been developed, although the efficacy of doxycycline and mTOR inhibitors such as rapamycin, in addition to the efficacy of an aromatase inhibitor, have been investigated in clinical trials.3 Occasionally, LAM can arise at extrapulmonary sites, such as the mediastinum, the retroperitoneum, or the lymph nodes. These lesions reveal a localized form of the disease, which is referred to as extrapulmonary lymphangioleiomyoma (E-LAM). Due to its rare occurrence and the variable, atypical locations of its associated lesions, E-LAM is often difficult to diagnose.4 Herein, we describe four cases of E-LAM, including the clinicopathological information of each case, and also review previous studies of patients with E-LAM.
MATERIALS AND METHODS
Four patients with E-LAM were identified at the Samsung Medical Center (Seoul, Korea) from 1995 to 2012. All cases were surgically resected. Complete clinicopathological data, including age, sex, E-LAM location, other clinical conditions, radiologic findings, size, follow-up period, and presence of TS were collected for each patient. Hematoxylin and eosin slides were reviewed to evaluate the histologic features of the E-LAM lesions in each patient. Immunohistochemical staining was also performed, including staining for smooth muscle actin (1:1,000, 1A4, Dako, Glostrup, Denmark), human malanoma black-45 (HMB-45; 1:80, Dako), the progesterone receptor (1:800, 16, Novocastra, Newcastle upon Tyne, UK), and p53 (1:10,000, BP53.12, Invitrogen, Carlsbad, CA, USA).
RESULTS
The clinicoradiological information for each patient is summarized in Table 1. All patients were female, and all were in the age range of 43 to 47 years old. Two patients (cases nos. 2 and 4) exhibited low-attenuated lesions (9 cm and 10 cm in diameter, respectively) in the para-aortic retroperitoneal area as assessed by computed tomography (CT) (Fig. 1A). The radiological conclusions from these scans were lymphangioma and lymphoma, respectively. One patient (case no. 1) exhibited a parametrial mass 6.5 cm in diameter, with a large number of additional lesions displaying an extensive mix of high and low attenuation by CT. In the remaining patient (case no. 3), E-LAM was incidentally detected in several lymph nodes of the right pelvis. This patient exhibited squamous cell carcinoma of the uterine cervix; thus, radical hysterectomy with bilateral pelvic lymph node dissection was performed. Two of the four patients also had accompanying P-LAM. The P-LAM exhibited characteristic radiologic features of well-defined, uniformly thin-walled cysts that were diffusely distributed throughout both lungs (Fig. 1B). Lung biopsies were not performed for these two patients. Furthermore, none of the patients exhibited any evidence of TS. The follow-up periods ranged from a third of a month to 95 months. All patients were alive at the end of follow-up.

Clinicoradiologic data from patients with extrapulmonary lymphangioleiomyoma in Korea
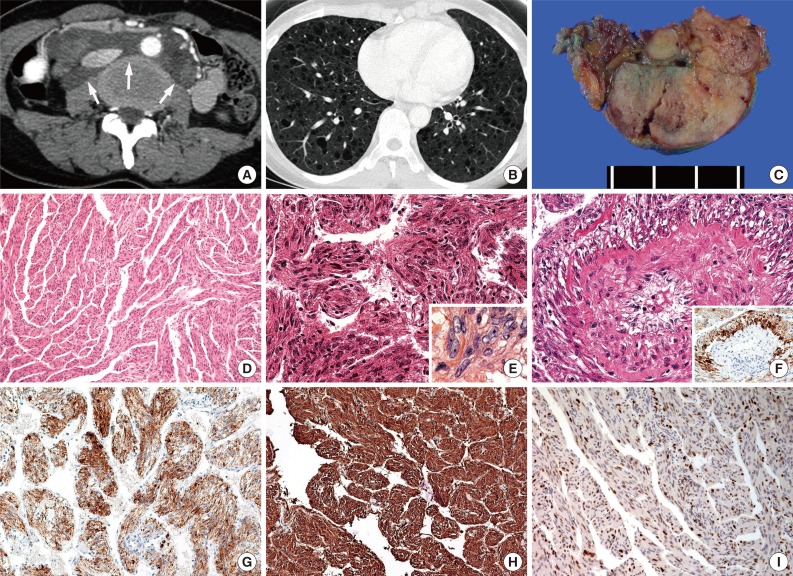
(A) Abdominal CT scan of case no. 4. A low-attenuated, para-aortic retroperitoneal mass is observed. (B) Chest computed tomography (CT) scan of case no. 4. Multiple well-defined, uniformly thin-walled cysts are observed diffusely distributed throughout both lungs. (C) Gross examination of multiple dissections revealing a soft, well-circumscribed, lobulated, ivory mass in case no. 2. (D) Microscope images of the masses observed in cases nos. 1 and 4. These masses are composed of bland-looking spindle cells and show trabecular bundles with anastomosing patterns (case no. 1). (E) Nuclear morphology of cases nos. 2 and 3. Nuclear crowding is observed, along with mild to moderate nuclear atypism and conspicuous nucleoli. (F) Perivascular epithelioid E-LAM (PEE) cell proliferation detected at the intratumoral area of case no. 2. The PEE cells stain more strongly positive for human melanoma black-45 (HMB-45) than do spindle tumor cells. (inset) (G) Positive staining for HMB-45 in spindle tumor cells (case no. 2). (H) Diffuse and strong positive staining for smooth muscle actin in spindle tumor cells (case no. 2). (I) Positive staining for the progesterone receptor in the nuclei of the tumor cells (case no. 1).
Gross examination of multiple dissections revealed soft, well-circumscribed, ivory and gray solid masses (Fig. 1C). The histopathologic findings were summarized in Table 2. Microscopically, these masses were composed of spindle cells, aggregated lymphocytes, and small vessels. The spindle cells formed anastomosing trabecular bundles with slit-like spaces, similar to a labyrinth (Fig. 1D). Several spindle cell areas showed vaguely palisading patterns. In cases nos. 1 and 4, relatively low cellularity was observed in these areas. The nuclei of the spindle cells exhibited bland-looking nuclear features, such as fine chromatin structures distributed openly and evenly, and inconspicuous nucleoli. In cases nos. 2 and 3, diffuse nuclear crowding was observed at low magnification. The tumor cells exhibited spindle-shaped or round nuclei, with a coarse chromatin pattern. Nuclear membrane wrinkling and conspicuous nucleoli were also identified in the tumor cells (Fig. 1E). These characteristics were also accompanied by cytoplasmic degeneration, with obscure cytoplasmic membranes, and amorphous pink material intermingled between the tumor cells. Perivascular epithelioid E-LAM (PEE) cell proliferation was detected particularly at the intratumoral area of case no. 2 (Fig. 1F). The proliferated PEE cells exhibited round or polygonal-shaped nuclei. The PEE cells were located around the vessels and extended up to the vessel adventitia. A coarse chromatin pattern and conspicuous nucleoli were identified in PEE cells. Case no. 3 did not exhibit any blood vessels near the tumoral lymph node lesions, and PEE proliferation was not observed, although several foci of lymphoid aggregation were observed. Mitosis or necrosis was not seen in any case.
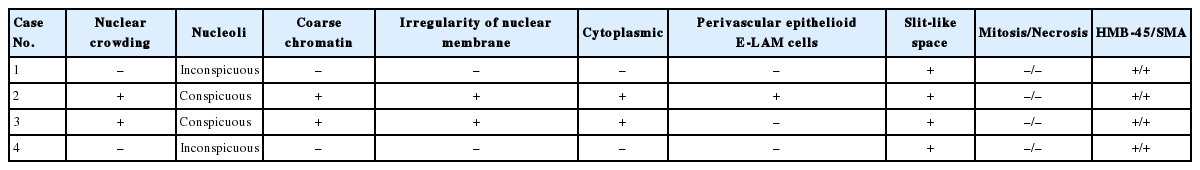
Histopathologic information from patients with extrapulmonary lymphangioleiomyoma
Immunohistochemical analysis revealed that the spindle-shaped and round E-LAM cells exhibited diffuse staining for HMB-45 (Fig. 1F, G). The PEE cells showed higher reactivity for HMB-45 compared with the surrounding the spindle shaped tumor cells. Furthermore, the spindle cells and the PEE cells showed diffuse staining for both smooth muscle actin and the progesterone receptor (Fig. 1H, I). All samples were negative for p53.
DISCUSSION
Lung tumors such as LAM, angiomyolipoma, and clear cell "sugar" tumors belong to the PEComa family.5 The World Health Organization has defined PEComa as "mesenchymal tumors composed of distinctive cells that show a focal association with blood vessel walls and usually express melanocytic and smooth-muscle markers." Perivascular epithelioid cells (PEC) are difficult to detect in normal tissue; thus, the role of PEC in these lung tumors is not yet well-established. The LAM cells in this study showed identical immunohistochemical staining for HMB-45 and smooth muscle actin as for PEC. These LAM cells have been shown to be related to lymphatic smooth muscle cells.6 The majority of LAM cases originate at the lung and manifest as diffuse cystic lesions. Microscopically, P-LAM cells are spindle-shaped or round and exhibit bland-looking nuclei. These cells form either a well-defined cystic wall or randomly distributed, proliferative foci. In contrast to P-LAM, which is manifested by diffuse lesions, E-LAM is manifested by localized lesions. Furthermore, E-LAM tumors are extremely rare, and the precise nature of these tumors is still being investigated.
Several cases of E-LAM have been reported. Matsui et al.6 described 22 cases of E-LAM, including the relevant clinicopathological information from each case. All 22 patients with E-LAM were female, and their ages ranged from 22 to 67 years old. Moreover, 19 of the 22 patients also exhibited P-LAM. The follow-up periods ranged from 1 to 13 years, and one patient died from P-LAM. The three primary locations of the E-LAM lesions were the posterior mediastinum, the upper retroperitoneum adjacent to the abdominal aorta, and the pelvic cavity. To the best of our knowledge, only two previous reports have described E-LAM cases in Korea.7,8 The first case of E-LAM was reported by Kim et al.7 in 2005, and a second case was reported by Han et al.8 in 2008. The six cases of E-LAM that have been identified in Korea, including the four present cases, are summarized in Table 1. All patients were female, and their ages ranged from 21 to 41 years old. The tumors were present in a sporadic form and were located in the pelvic cavity in four patients and in the para-aortic retroperitoneum in two patients. Three patients also had accompanying P-LAM. The youngest patient died from P-LAM 6 months after the initial operation. Currently, E-LAM is very difficult to diagnose by radiology in the absence of accompanying P-LAM.
Differential diagnoses performed with radiological techniques should include lymphoma, schwannoma, paraganglioma at the para-aortic area, and metastatic tumors in the lymph nodes. The two present cases with para-aortic masses appeared to be lymphoma and paraganglioma by radiological examinations. Pathologically, determining the extent of immunoreactivity for HMB-45 and smooth muscle actin is helpful in differential diagnosis between schwannoma, leiomyoma, leimyosarcoma, and paraganglioma. The distinctive structural pattern of E-LAM lesions, which consist of an anastomosing trabecular bundle with slit-like spaces similar to a labyrinth, is also a valuable clue in differential diagnosis from PEComa. Using intraoperative frozen biopsies, the discrimination of paraganglioma and metastatic leiomyosarcoma from E-LAM was extremely difficult, and may not actually be possible.
The majority of E-LAM cases are also accompanied by P-LAM.6 Out of the six cases of E-LAM in Korea, including the four cases in our study, three patients exhibited concurrent P-LAM and E-LAM. It is currently vigorously contested whether E-LAM and/or P-LAM lesions are metastatic or not. Hayashi et al.9 described metastatic potential in the LAM lesions of 10 patients. These authors suggested that the primary site of LAM may be the uterus or the pelvic cavity near the uterus, thus raising the possibility that the P-LAM lesions were metastatic lesions originally derived from the uterus. They introduced PEE cells as involvement of vascular wall of LAM cells. Out of the nine cases of uterine LAM, seven exhibited involvement of the vascular wall; however, intravascular involvement was not detected and endothelial cells were preserved in all cases.9 In our study, case 2 exhibited PEE cells with preserved endothelial cells and no intravascular involvement. And this case of E-LAM was not accompanied by P-LAM. Our study is the first to report the finding that PEE cells exhibit stronger staining for HMB-45 compared with spindle LAM cells. To accurately evaluate the degree of metastasis of LAM lesions, it is vital to determine whether the PEE cells represent an invasion or not. To date, no definitive evidence has been obtained regarding whether LAM lesions exhibit invasion or metastasis.
Cases 2 and 3 revealed cytologic atypism, compared with cases 1 and 4, which did not. The cases of E-LAM that exhibited cellular atypism (cases 2 and 3) were not accompanied by P-LAM. Considering case 2 showed both PEE and cellular atypism, the cellular atypism of E-LAM may be a valuable feature for determining whether or not the E-LAM is accompanied by PEE cells. Case 3 showed cellular atypism; however, PEE cell was not detected in this case. However, the opportunity to evaluate the presence of PEE in case 3 was somewhat limited, since no medium-sized vascular structures were observed in the intratumoral area. In PEComa, variable degrees of tumor size, infiltration, nuclear grade, cellularity, mitotic activity, necrosis, and vascular invasion are considered to indicate malignancy.10 PEComa with nuclear pleomorphism was considered to have uncertain malignant potential by Folpe et al.10 However, the criteria for nuclear atypism in E-LAM have not yet been defined. Further clinicopathological correlating study including more cases with nuclear atypism, are needed.
In addition, based on the conclusions of several previous reports,11,12 we immunostained for p53 in the key slides of all our cases in an attempt to identify possible differences between atypical nuclei and bland nuclei. However, none of the slides in our study stained positive for p53.
In conclusion, here we report four rare cases of E-LAM in our hospital. For females in whom para-aortic or pelvic masses are observed, E-LAM should be considered in the differential diagnosis. Further clinical and pathological evaluation of E-LAM with nuclear atypism is necessary.
Notes
No potential conflict of interest relevant to this article was reported.