Follicular lymphoma: updates for pathologists
Article information

Abstract
Follicular lymphoma (FL) is the most common indolent B-cell lymphoma and originates from germinal center B-cells (centrocytes and centroblasts) of the lymphoid follicle. Tumorigenesis is believed to initiate early in precursor B-cells in the bone marrow (BM) that acquire the t(14;18)(q32;q21). These cells later migrate to lymph nodes to continue their maturation through the germinal center reaction, at which time they acquire additional genetic and epigeneticabnormalities that promote lymphomagenesis. FLs are heterogeneous in terms of their clinicopathologic features. Most FLs are indolent and clinically characterized by peripheral lymphadenopathy with involvement of the spleen, BM, and peripheral blood in a substantial subset of patients, sometimes accompanied by constitutional symptoms and laboratory abnormalities. Diagnosis is established by the histopathologic identification of a B-cell proliferation usually distributed in an at least partially follicular pattern, typically, but not always, in a lymph node biopsy. The B-cell proliferation is biologically of germinal center cell origin, thus shows an expression of germinal center-associated antigens as detected by immunophenotyping. Although many cases of FLs are typical and histopathologic features are straightforward, the biologic and histopathologic variability of FL is wide, and an accurate diagnosis of FL over this disease spectrum requires knowledge of morphologic variants that can mimic other lymphomas, and rarely non-hematologic malignancies, clinically unique variants, and pitfalls in the interpretation of ancillary studies. The overall survival for most patients is prolonged, but relapses are frequent. The treatment landscape in FL now includes the application of immunotherapy and targeted therapy in addition to chemotherapy.
Follicular lymphoma (FL) is an indolent B-cell malignancy usually involving a lymph node with a morphologic appearance resembling follicular lymphoid structures. FL cells arise from germinal center B-cells of the lymphoid follicle (centrocytes and centroblasts) [1]. The biologic and histopathologic spectrum of FL is broad. In most cases, at least partial follicular (nodular) distribution is identified with varying degrees of diffuse neoplastic growth [2].
In 2017 World Health Organization (WHO) classification, grading of FL is based on the cytologic composition of neoplastic follicles and the number of centroblasts identified, on average, in at least 10 high power fields. FLs express germinal center B-cell markers by immunohistochemistry (IHC) [1].
Obsolete names for FL are Brill-Symmers disease, nodular lymphoma (in Rappaport classification), centroblastic/centrocytic lymphoma (Kiel classification), and follicular center lymphoma (in REAL classification) [2-5].
FL is the second most common non-Hodgkin lymphoma, comprising about 20%–25% of all new non-Hodgkin lymphoma diagnoses in Western countries, and about 5% of all hematologic neoplasms. The incidence of FL differs among geographic regions and ethnic groups. Age-standardized incidence is about 2–4 per 100,000 person-years, and male to female ratio is variable (1:1.7 in most series). The median age ranges 60–65 years [6].
About 80% of all FLs are systemic nodal tumors. Peripheral cervical and inguinal lymph nodes are the more frequently affected sites, although central lymph nodes, including abdominal and thoracic, can also be involved. Extranodal sites that are commonly involved include bone marrow (BM), spleen, liver, and peripheral blood [7].
ETIOPATHOGENESIS
Biologic abnormalities that promote the development of FL can be broadly summarized as occurring in three stages of B-cell development: (1) BM events, (2) germinal center events, and (3) post-germinal center events.
BM events
BM precursor B-cells, usually in the pre- or pro-B-cell stage, acquire t(14;18)(q32;q21) IGH/BCL2 because of repair failure during V(D)J recombination. The resulting overexpression of BCL2, an anti-apoptotic protein, promotes survival and discourages apoptosis of B-cells as they later mature during the germinal center reaction [8].
Germinal center events
B-cells harboring the t(14;18)(q32;q21) translocation retain germinal center functionality (e.g., BCL6-mediated pathways), and undergo somatic hypermutation and class switch recombination of immunoglobulin genes initiated by activation-induced cytidine deaminase with retention of IgM/IgD surface expression. This last phenomenon is known as the allelic paradox and promotes proliferation and survival pathways in malignant Bcells [9].
Post-germinal center events
Additional chromosomal alterations and mutational abnormalities occur, promoting the pre-lymphomatous t(14;18)+ cell into bonafide lymphoma cells. Reentry of a subset of BCL2+ memory B-cells to the germinal center also occurs [10].
In addition to the t(14;18)(q32;q21) translocation and BCL2 mutation, FL is characterized by additional mutational abnormalities most commonly in chromatin modifiers, B-cell receptor signaling pathways, cell cycle regulation, transcription factors, and immune evasion genes [10-13]:
- Chromatin modification (KMT2D, EZH2, CREBBP, ARID1A, MEF2B, EP300): inactivating mutations of KMT2D, CREBBP, ARID1A, MEF2B, and EP300 and gains of the function of EZH2.
- B-cell receptor signaling (CARD11, IgHV, IgLV, TNFRSF14): inactivating mutation of TNFRSF14 and gain of function mutation in CARD11, IgHV, IgLV.
- Cell cycle regulation (RB1, CDK4): deletion of RB1 and gain of function mutation in CDK4.
- Transcription factors (FOXO1, MEF2B, BCL6): inactivating mutation of MEF2B and gain of function mutation of FOXO1 and BCL6.
- Tumor suppressor and immune evasion (EPHA7, TNRSF14, CREBBP): deletion/inactivating mutation of EPHA7, TNRSF14, and CREBBP.
- Activation of JAK-STAT signaling (STAT6): gain of function mutation in STAT6.
The etiology of FL is mainly unknown. t(14;18)(q32;q21) IGH/BCL2 alone is not sufficient to cause lymphoma [14]. Some essential factors which seem to play a contributing role are family history and inherited/genetic susceptibility (especially first-degree relatives) and environmental factors (such as exposure to pesticides and herbicides) [15,16].
Clinical features
The median age of patients with FL is the sixth decade [1]. Clinical presentation of FL is most commonly that of enlarged lymph nodes, frequently in the neck or abdomen. FL is a localized disease in about 10% to 20% of cases. The vast majority of FLs present with widespread nodal involvement (~80%) and advancedstage disease (stages III–IV) at the time of diagnosis [7]. Some cases follow a chronic relapsing course. A subset progresses rapidly and transforms to aggressive lymphomas such as diffuse large B-cell lymphomas, double-hit large B-cell lymphomas, and lymphoblastic leukemia/lymphoma [17].
FL can also relapse as classic Hodgkin lymphoma, which is clonally related to the antecedent FL and with Hodgkin lymphoma cells also harboring the t(14;18) (q32;q21) translocation [18].
Most patients with FL are mainly asymptomatic. Symptomatic presentations may include fatigue, fever or night sweats, weight loss, or recurrent infections. Tissue biopsy (lymph node/extranodal sites), most frequently in the form of needle biopsy (both core needle biopsy and fine-needle aspiration biopsy), and occasionally as excisional biopsies, are the most frequent diagnostic materials. Peripheral blood and BM biopsy are usually performed for staging purposes [10].
Abnormal laboratory findings are uncommon. Leukemic phase FL is identified in <5% of patients [19]. An increase in lactate dehydrogenase (LDH) and β2-microglobulins are present in about 15% of patients [20].
Rarely, FL is found outside lymph nodes. Extranodal FL can cause a variety of symptoms depending on its location. For example, in case of BM involvement, anemia is present (about 10% of patients with FL), while leukopenia/thrombocytopenia is rarely seen [19,21]. Involvement of mucosa-associated sites may be asymptomatic or present with symptomatology related to the involved disease site.
HISTOLOGIC FINDINGS
The diagnosis of FL is made histologically in tissue sections obtained from surgically excised lymph nodes or, more frequently, a needle biopsy (both core needle biopsy and fine-needle aspiration biopsy).
Gross examination of involved lymph nodes shows a vaguely nodular pattern in the cut section with bulging.
Microscopic examination shows partial or complete effacement of the lymph node architecture with numerous, similarly sized, nonpolarized neoplastic follicles with attenuated or absent mantle zones, typically present in a back-to-back fashion in involved sites (Fig. 1). The neoplastic follicles of FL may also contain many reactive T-cells and follicular dendritic cells (FDCs), but tingible body macrophages are usually few or absent. FL is typically composed of centrocytes (small and large cleaved B-cells) and larger centroblasts (large noncleaved B-cells).
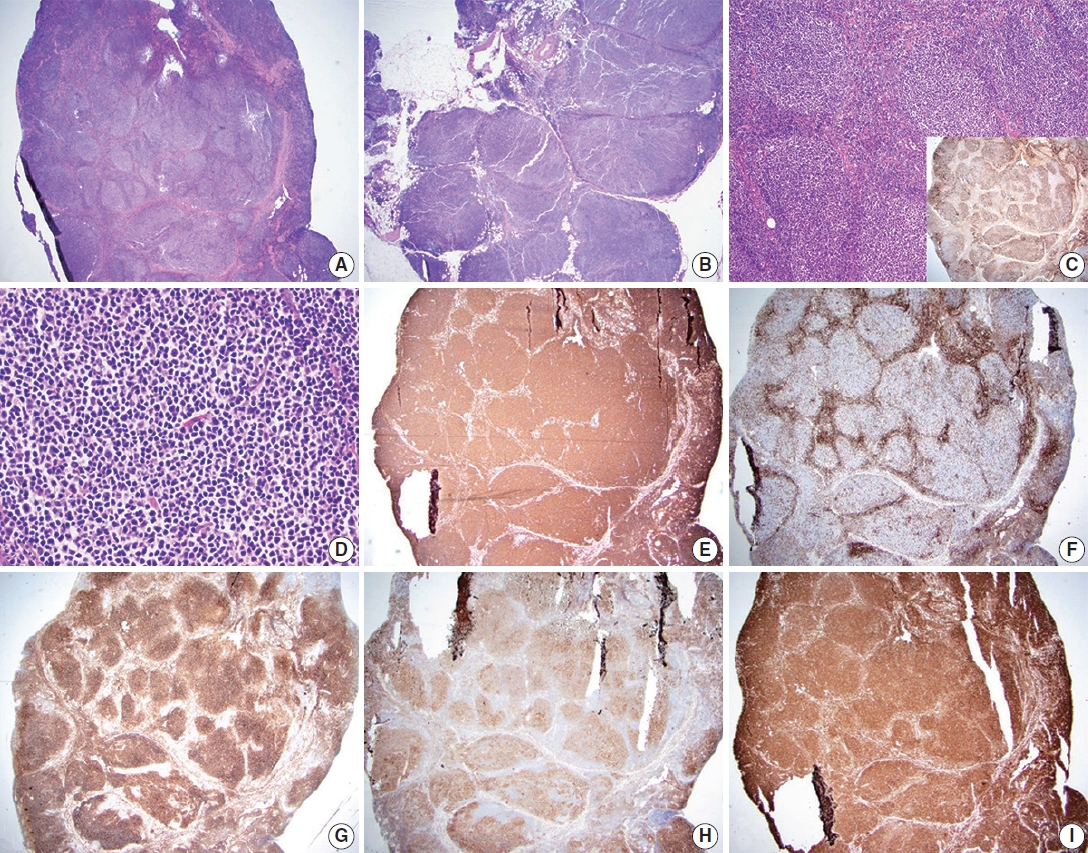
Follicular lymphoma, low-grade. A representative case of follicular lymphoma, low-grade. H&E-stained excisional biopsy (A–D) and immunohistochemical stains (C inset, E–I) show classic morphology of follicular lymphoma cells, with increased, monotonous appearing neoplastic follicles in an excisional biopsy of the lymph node. The borders of the follicles are ill-defined and lack well-preserved mantle zones. Foci of sclerosis are identified (A). The neoplastic follicles are expansile and arranged in a back-to-back fashion. The neoplasm extends into perinodal fat (B) and has attenuated to absent mantle zones (C). Immunostain for CD21 highlights follicular dendritic cell meshworks within neoplastic follicles, which is useful in establishing the presence of lymphoid follicles (C, inset). The neoplastic follicle comprises numerous centrocytes and fewer centroblasts, compatible with grade 1–2 of 3 (D). Immunostain for CD20 highlights B lymphocytes in neoplastic follicles and interfollicular (diffuse) areas (E). Immunostain for CD3 highlights reactive T-cells in follicular lymphoma. The pattern of CD3, accumulating around neoplastic follicles, can be used to highlightthe nodulardistribution of lymphoma cells (F). Immunostain for CD10 confirms that the neoplastic cells are of germinal center origin (lymphoma cells are positive within neoplastic follicles). Scattered interfollicular neoplastic cells are weakly stained with CD10. The reactivity is stronger in germinal centers than in interfollicular regions (G). Immunostain for BCL-6 highlights neoplastic lymphoma cells of germinal center origin within neoplastic follicles (H). Immunostain for BCL2 is positive in neoplastic Blymphocytes (I).
Grading of FL is based on the number of centroblasts per high-power microscopy field (×40 objective, 0.159 mm2) [1]. Distinguishing between grades 1 and 2 (containing up to 15 centroblasts per field) is not recommended at this time. Grade 3 cases have >15 centroblasts per field and are further subdivided to 3A (centrocytes present), 3B (solid aggregates of centroblasts with no or very rare intervening centrocytes) (Fig. 2). Grade 3A (or 3B) FL with diffuse growth containing >15 centroblasts per high-power field should be classified as diffuse large B-cell lymphoma (DLBCL) according to the WHO system (Table 1) [1].
Follicular lymphoma, high-grade morphology. A representative case of follicular lymphoma, high-grade (grade 3B). H&E-stained excisional biopsy (A, B). The neoplastic follicles are composed of a homogeneous population of large lymphoma cells. High power magnification shows a neoplastic follicle of FL, grade 3B. Most cells in this follicle are large centroblasts without intervening centrocytes.

Follicular lymphoma grading, pattern, immunohistochemical and cytogenetic findings
WHO classification recommends documenting the proportion of the neoplasm that is present in a follicular versus diffuse distribution, as follows: follicular pattern (>75% of the sample has a follicular pattern); follicular and diffuse pattern (25%–75% of the specimen has a follicular pattern); focally follicular/predominantly diffuse (<25% of the specimen has a follicular pattern); and a diffuse pattern (absence of follicular areas) [1].
Based on these criteria, a neoplasm with a purely follicular pattern is considered FL, even if composed of centroblasts alone. For patients with low-grade FL, the tumor pattern of diffuse has no prognostic importance. Still, the possibility of sampling error in small biopsies should be considered or noted in the report. An area of increased subjectivity is when biopsies are small and show limited regions of diffuse pattern with grade 3A morphology. In these cases, especially if associated with low-grade FL in most specimens and clinical findings support low-grade FL, it is critical not to overcall that diffuse area as DLBCL [22].
FL of the usual type can show a wide range of morphologic variability, but are still classified as FL. Distinct from this, there are also WHO-defined FL variants which are also FL but have consistent clinicopathologic and biologic nuances that separate them from usual FL. In terms of morphologic variability in FL, some cases of FL, especially when neoplastic follicles invade beyond the lymph node capsule, particularly in retroperitoneal and mesenteric sites, can be associated with diffuse and prominent sclerosis, often associated with blood vessels. FL of the usual type may also show scattered Hodgkin-like cells, which must be distinguished from classic Hodgkin lymphoma and collision tumors with both FL and classic Hodgkin lymphoma, usually accomplished by careful and extensive immunophenotyping. FL can also have Castleman-like features, including concentric mantle zone cells, hyalinization and regression of follicles, and interfollicular vascular proliferation with penetrating vessels creating lollipop lesions. These cases may mimic hyaline vascular Castleman’s disease to the point that the FL is not identified, creating a diagnostic pitfall [23]. In the floral variant of FL, neoplastic follicles are irregular in shape and surrounded by expanded, prominent mantle zone lymphocytes which penetrate neoplastic follicles. This variant FL resembles the non-neoplastic entity progressive transformation of germinal centers and may also mimic other lymphomas, including marginal zone lymphoma (with follicular colonization) or nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) [24,25]. Signet ring cell FL is a variant in which tumor cells have clear, vacuolated cytoplasm and an eccentric nucleus and should be distinguished from carcinoma cells. The vacuoles are composed of intracytoplasmic immunoglobulin deposits [26].
FL variants include in situ follicular neoplasia, duodenal-type FL, diffuse variant FL, and testicular FL. In situ follicular neoplasia is diagnosed when lymph node biopsies show overall normal histologic findings with preservation of nodal architecture. Still, abnormal, bright BCL2-positive B-cells are identified within lymphoid follicles [1]. These BCL2-positive B-cells are confined to follicles and represent colonization of pre-existing germinal centers by monoclonal BCL2-rearranged B-cells [27,28]. Because a subset of these patients will have FL in other sites at the time of diagnosis of in situ follicular neoplasia, these patients should be staged. Approximately 5% of these patients will subsequently develop FL or DLBCL [29]. Further supporting that these lesions are biologically in situ neoplasms, sequencing studies performed in these selected cells show mutation abnormalities similar to those seen in FL but at lower variant allele frequency. Moreover, retrospective analysis of previously removed lymph nodes in patients diagnosed with FL can identify in situ FL in most of these patients [30,31].
As opposed to in situ follicular neoplasia, lymph nodes may show partial involvement by FL. Both neoplastic and reactive follicles are present in these cases, and lymph node architecture is partially effaced. These cases are still classified as FL, not in situ lesions, and the presence of only partial nodal involvement is associated with lower stage, and better prognosis, given adequate sampling [32].
Duodenal-type FL are FL that arise at extranodal, mucosal sites within the small bowel, usually the second portion of the duodenum (Fig. 3). These neoplasms do not typically pose diagnostic challenges since the histopathologic features are typical of low-grade FL. This variant is important to recognize clinically since it occurs in younger patients and remains localized in nature [33]. Therefore, it is amenable to localized radiation alone and generally does not require systemic therapy. Biologically this variant FL is intriguing in that it shows features overlapping between FL and extranodal marginal zone lymphoma. Like usual FL, duodenal-type FLs have BCL2 translocations, somatically hypermutated immunoglobulin genes, and frequent mutations in KMT2D, CREBBP, and TNFRSF14 [34]. However, these lesions additionally show features that overlap with those of extranodal marginal zone lymphoma, including restricted usage of immunoglobulin heavy chain variable region, suggesting development in the context of antigen stimulation [35].
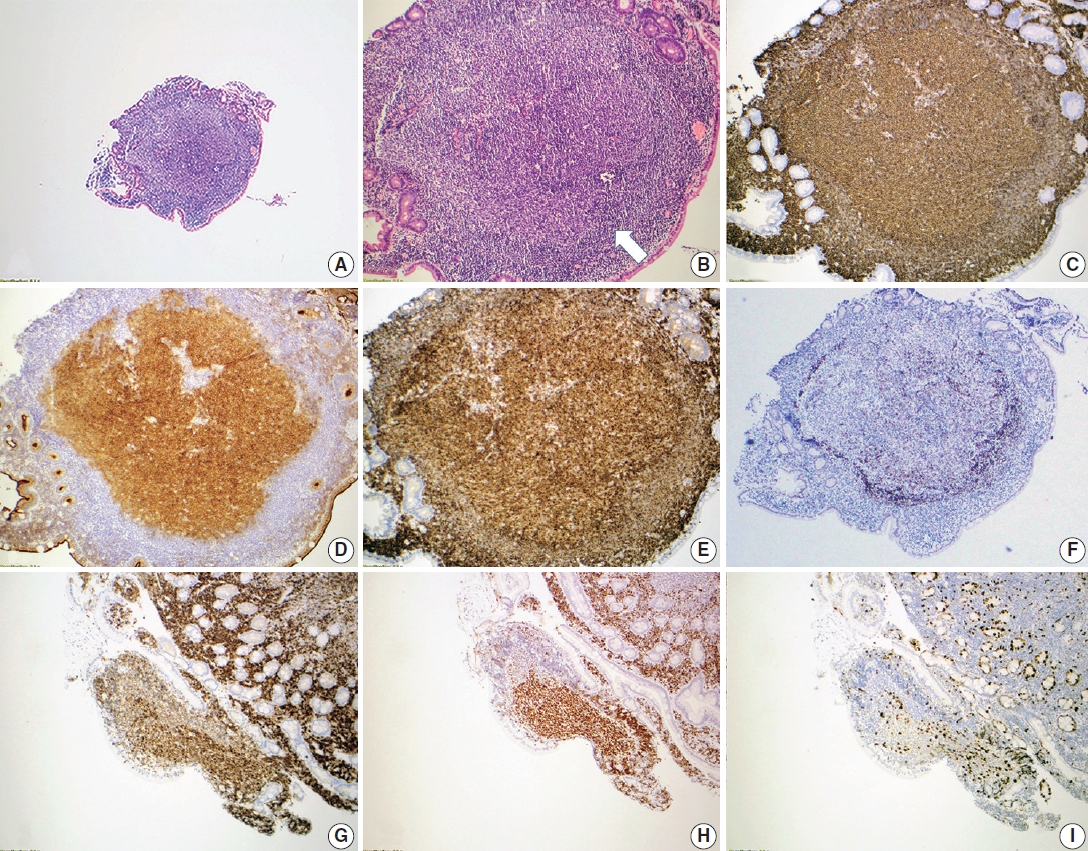
Duodenal-type follicular lymphoma. A representative case of duodenal-type follicular lymphoma. H&E-stained biopsy (A, B) and immunohistochemical stains (C–I) show intramucosal follicular lymphoma, grade 1, forming polyps in the mucosa of the small intestine (A). The neoplastic follicles are partially surrounded by a thin layer of mantle cells (arrow) (B). Neoplastic lymphocytes are positive for CD20 (C) with the germinal center marker CD10 (D), and BCL6 (H) co-expression. CD10 also highlights the intestinal absorptive epithelium, which is internal control. The neoplastic lymphocytes co-express BCL2 (E, G), supporting follicular lymphoma diagnosis. CD21 confirms that the follicular dendritic meshwork is located at the periphery of the neoplastic follicle (F). Ki-67 proliferation index is not increased (I).
The diffuse variant of FL has clinical, immunophenotypic, and molecular genetic differences from typical nodal FL. This variant FL typically presents at nodal sites, usually inguinal lymph nodes, as a large mass [36,37]. However, as opposed to the usual FL, this variant is frequently localized without systemic involvement. Histologically, this variant may be challenging to recognize, given that the majority of the neoplasm is distributed in a diffuse pattern with only focal and usually small micronodular foci. Cytologically, lymphoma cells have typical centrocytic and centroblastic morphologic features and typically express CD10 and other germinal center B-cell markers. Diffuse expression of CD23 is consistently identified. Like usual FL, these neoplasms frequently show 1p36 chromosomal abnormalities and/or TNFSRF14 and CREBBP mutations. However, unlike usual FL, these neoplasms lack the BCL2 translocation harbor STAT6 mutations.
The testicular variant of FL is rare. This neoplasm was initially identified in children but has also rarely been reported in adults [38]. These neoplasms have several features similar to pediatric type FL. Histologically, these neoplasms have high-grade cytology (grade 3A or 3B histology), yet they are usually localized and associated with a good prognosis. Neoplastic cells in this variant do not express BCL2 protein and lack the BCL2 translocation, similar to pediatric type FL.
Needle biopsy of BM is performed as part of staging procedures in patients with FL. In FL, focal or extensive BM involvement is found in most patients [39]. The most frequent pattern of involvement is paratrabecular aggregates of lymphoma cells, with or without interstitial or diffuse patterns. The pure follicular (nodular) pattern in BM is present in about 5% of cases with BM involvement [40].
The liver and spleen are commonly involved in FL. In the liver, FL usually involves the portal tracts. However, large nodules in parenchyma are present when the spleen is extensively involved by lymphoma. In the spleen, the white pulp is preferentially involved with two predominant patterns: Expansion of the white pulp in most cases vs. relatively preserved white pulp architecture less frequently [41,42].
Fine-needle aspirations typically show variable mixtures of centrocytes and centroblasts. In comparison to reactive follicular hyperplasia, tingible body macrophages are rare or absent (Fig. 4). Centrocytes are small to large, have angulated nuclei, dense chromatin, and scant cytoplasm. Centroblasts are large cells with oval nuclei, vesicular chromatin, 1–3 nucleoli, moderate cytoplasm and are >three times the size of lymphocytes. Similar morphologic features might be seen in FDCs. However, FDCs have large round nuclei, dispersed and even nearly clear chromatin, single eosinophilic nucleolus, and indistinct cytoplasm [43].
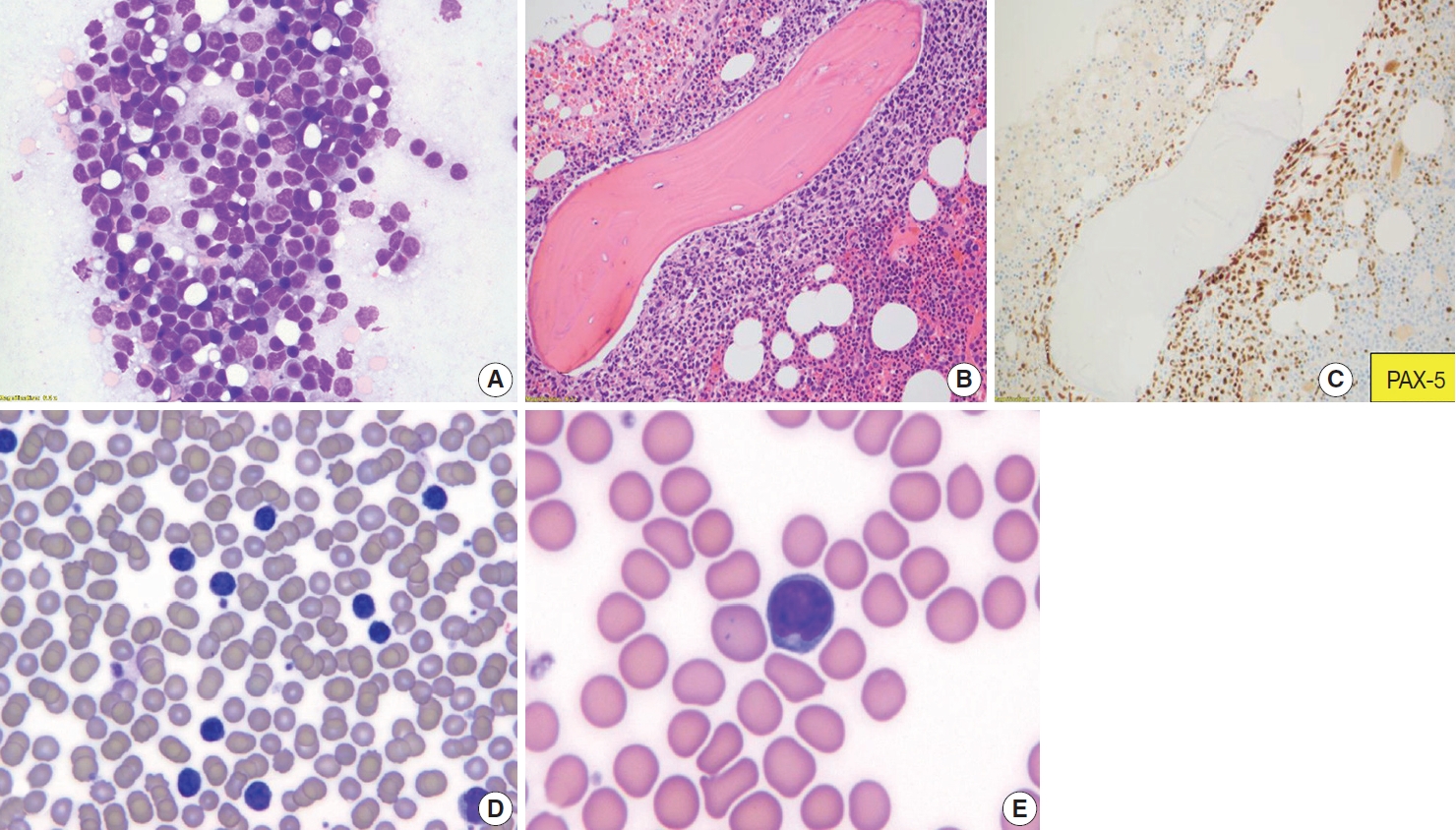
Follicular lymphoma, fine-needle aspirate smear. Paratrabecular pattern of involvement in the bone marrow and peripheral blood smear involvement. Wright-Giemsa-stained smears (A, D, E), H&E-stained slide (B), and immunohistochemical stain (C). Fine-needle aspiration of a lymph node from a patient with FL, grade 1–2, demonstrates a mixture of centrocytes and centroblasts (A). Bone marrow core biopsy specimen involved by follicular lymphoma is shown. The neoplasm has a paratrabecular pattern of distribution (B). PAX-5 highlights neoplastic B-cells (C). Peripheral blood smear from a patient with follicular lymphoma demonstrates leukemic involvement by centrocytes (D) with deeply cleaved nuclei (E).
IMMUNOHISTOCHEMICAL AND ANCILLARY STUDIES
By IHC, FL cells express pan-B-cell markers (CD19, CD20, CD22, CD79, PAX5) and monotypic surface Ig, most commonly IgM with or without IgD [1]. Germinal center markers, including CD10, BCL6, HGAL, LMO2, STMN1, GCET, MEF2B, etc., are variably expressed by FL cells [44-46]. BCL2 is positive in 85%–90% of FL grades 1 and 2 [46]. BCL2 and CD10 show decreased intensity and frequency of staining as the grade of FL increases (Table 1).
A subset of FL cases is truly negative for BCL2, especially in grade 3B [47-49]. These tumors also usually do not have the BCL2 translocation of FL and may express MUM1 and cytoplasmic immunoglobulin, thus appearing to have biology more similar to post-germinal center B-cells. However, a subset of cases is falsely negative for BCL2 expression, which may be due to a point mutation in BCL2 that blocks binding of the BCL2 clone 124, which is used commonly in clinical IHC. Other anti-BCL2 antibodies such as clone E17 or SP66 will be positive [50].
FL cases are negative for T cell markers CD2, CD3, CD4, CD5, CD7, CD8, and are usually negative for CD43 and cyclin D1 [1,51]. A small subset of cases is positive for CD5, including a subset of instances of the floral variant of FL [52].
The morphologic appreciation of follicular patterns in FL is usually adequate. Still, it can be supported by identifying FDC networks underlying the follicular aggregates, which are generally positive for CD21, CD23, and/or CD35 [22]. Another clue for a nodular pattern in FL is the accumulation of reactive T-cells at the periphery of the nodules. CXCL13 is another FDC-associated IHC marker that tends to be positive in most FL cases with FDCs when negative for other FDC-associated IHC markers [53].
Ki-67 assesses the proliferation rate of follicular lymphomas, and the Ki-67 proliferation index correlates with FL grade. Most low-grade FL has proliferative rates <20%. However, about 20% of low-grade FL have a high proliferation rate (>30%). These follicular lymphomas appear to behave more aggressively, similar to grade 3A or grade 3B FL. We report these cases as FL grade 1 to 2 with a high proliferation fraction. We include diagnostic comments indicating that these cases may be more clinically aggressive than typical low-grade FL [54]. It is recommended to assess Ki-67 in follicles. However, if interfollicular areas are more extensive than follicular ones, we estimate the Ki-67 proliferation based on an average of the entire neoplasm [22]. FLs with polarized follicles may have higher Ki-67 proliferative rates present in the dark zone of the follicle, and this should not be interpreted as a high proliferation index in FL [22].
Peripheral blood is commonly involved (at a low level) in ~90% of FL cases. This feature can be detected by flow cytometry or molecular methods if not observed easily in peripheral blood smear. Absolute lymphocytosis with a high count is present in 5%–10% of cases [19]. Neoplastic cells are typically small to intermediate in size with highly indented nuclei, known as buttock cells.
The typical immunophenotype of FL as identified by flow cytometry is CD19+ (dim), CD20+ (bright), CD10+ (uniform), CD5–, CD23–, CD200–, CD11c–, with surface expression of restricted immunoglobulin κ-chain or immunoglobulin λ-chain (Fig. 5).

A representative case of the flow cytometric immunophenotype of follicular lymphoma. Flow cytometry immunophenotyping of a lymph node fine-needle aspirate specimen from a patient with follicular lymphoma confirms that the lymphoma cells, gated by expression of CD45, co-express CD20 and CD10, with surface light chain restriction for lambda.
Conventional cytogenetic analysis can detect t(14;18)(q32;q21) IGH-BCL2 in up to 90% of FL cases (Fig. 6) [55].
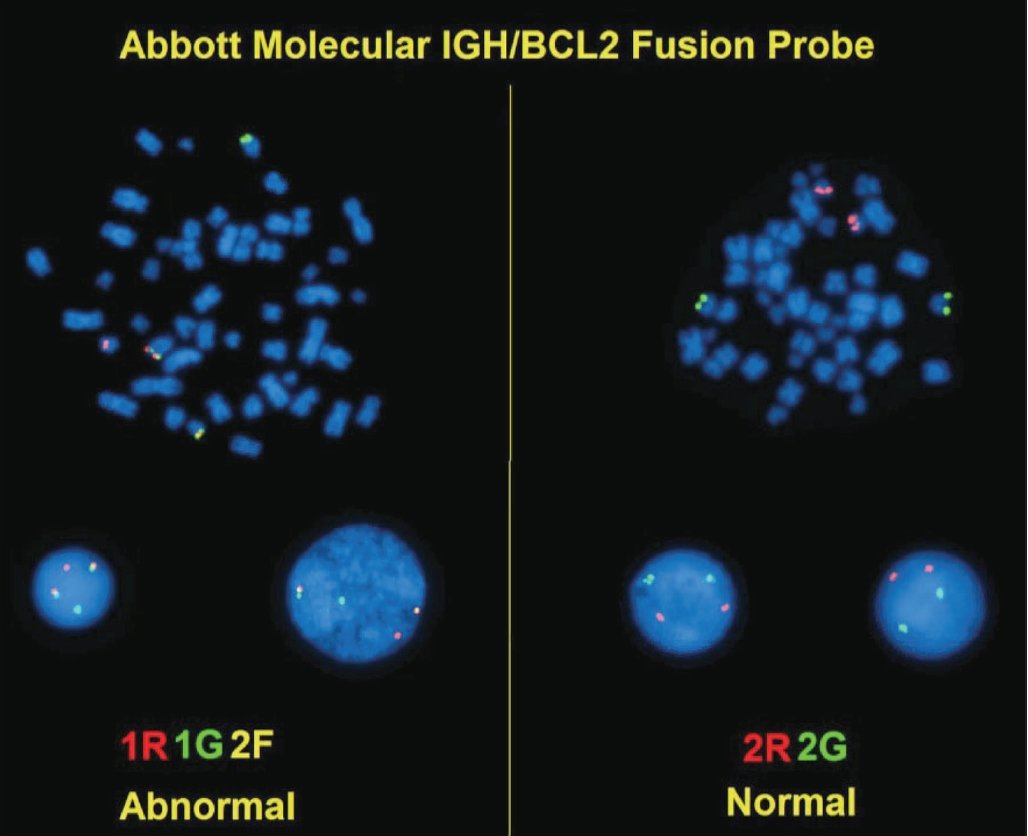
IGH/BCL2 dual-color fluorescent in-situ hybridization (FISH). FISH on a fixed, paraffin-embedded tissue section of follicular lymphoma using dual-fusion probes for BCL2 (red) and IGH (green). The t(14;18)(q32;q21) IGH/BCL2 fusion gene is a yellow signal.
Fluorescent in-situ hybridization (FISH) is more sensitive than PCR-based approaches in detecting IGH-BCL2 due to variation in breakpoint regions [56]. Rare cases of FL have the juxtaposition of the Ig light chain promotors to BCL2: t(2;18)(p11;q21) and t(18;22)(q21;q11) [57].
The absence of a BCL2 rearrangement in a suspected low-grade FL is unusual. In contrast, the absence of such a rearrangement in grade 3 cases should not be interpreted as evidence against a diagnosis of follicular lymphoma, particularly in grade 3B FL.
Loss of 1p36, which contains TNFRSF14, is common in FL [11,13]. 3q27 BCL6 rearrangement or amplification is present mainly in FL 3B. In the absence of BCL2 rearrangement in grade 3 cases, FISH studies for BCL6 rearrangements can be performed.
MYC rearrangement/activation of MYC is rare in FL (<5%). In the absence of histologic transformation, these cases are still called FL and are not classified as high-grade B-cell lymphoma. More extensive studies are needed to evaluate if MYC-rearrangement in FL has prognostic significance [58,59]. However, some of the FL with MYC-rearrangement are associated with transformation to DLBCL. These cases are categorized as double-hit lymphoma, in the category of high-grade B-cell lymphoma, if transformed and have BCL2 or BCL6 translocation in addition to MYC-rearrangement [60]. Other genetic alterations associated with transformation to DLBCL include the inactivation of TP53 and CDKN2A [61,62].
Frequent genetic alterations in FL include: BCL2, KMT2D/MLL2, EPHA7, EZH2, BCL6, CREBBP, and TNFRSF14 mutations. Less common genetic alterations are ARID1A, CARD11, TNFAIP3, MYC, FOXO1, and TP53 mutation/alteration. Comparative genomic hybridization has detected gains in 1, 2p15, 6p, 7p, 7q, 8q, 12q, 18p, 18q, X chromosomes and losses in 1p36, 3q, 6q, 9p, 10q, 11q, 13q, 17p chromosomes [1,10,63].
Finally, monoclonal B-cell populations in FL can be detected by monoclonal Ig heavy and light chain gene rearrangements detected by BIOMED-2 primer sets in multiplex polymerase chain reaction [64,65].
DIFFERENTIAL DIAGNOSIS
Reactive follicular hyperplasia
In reactive follicular hyperplasia, follicles are primarily located in the cortex, are more widely separated, show variation in size and shape, the polarization of germinal centers into light and dark zones (higher proliferation), frequent mitoses, tingible body macrophages in germinal centers, and sharply demarcated mantle zones. There is usually no evidence of monoclonality by Ig rearrangement studies, and t(14;18)(q32;q21) is not identified [66]. Flow cytometry evidence of monotypic light chain expression, uniform expression of CD10, decreased intensity of CD19, CD20, and CD38, are also features that would support the diagnosis of FL over reactive follicular hyperplasia. However, rare cases of reactive lymph nodes can show monotypic light chain expression among B-cells, especially in children [67-69].
Progressive transformation of germinal centers
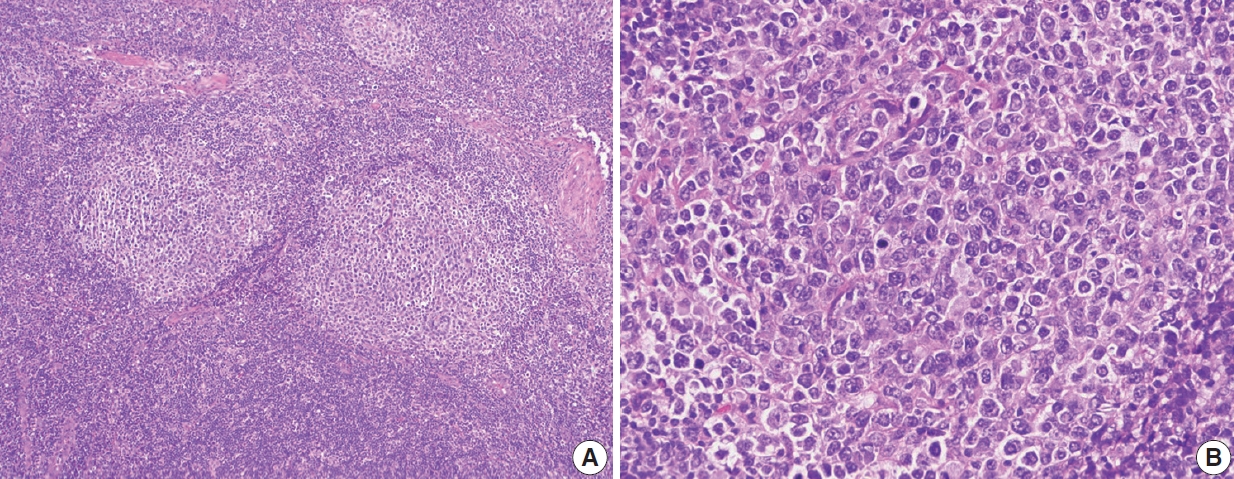
Nodules are 3–5 times larger than background reactive follicles in the progressive transformation of germinal centers. It may be hard to separate this entity from the floral variant of FL on morphology (Fig. 7). By IHC, germinal center B-cells are negative for BCL2 in the progressive transformation of germinal centers, and there is no evidence of monoclonality or t(14;18)(q32;q21)[25].
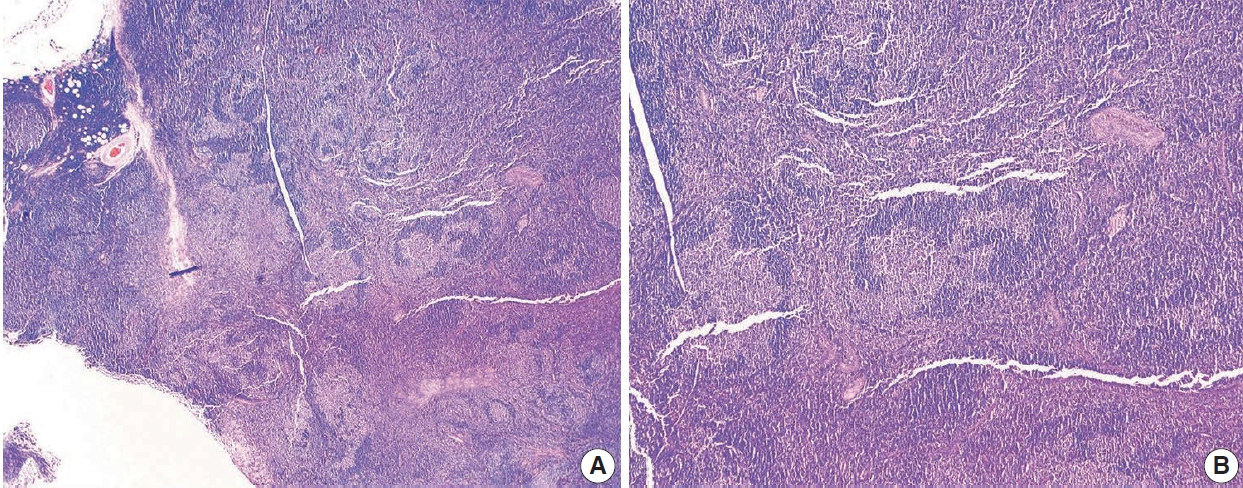
Floral variant of follicular lymphoma. H&E-stained excisional biopsy (A, B) shows the morphology of follicular lymphoma with large and lobulated follicles, in some areas resembling flower petals (A). The darker mantle zone lymphocytes are infiltrating the pale follicular lymphoma cells (B).
Castleman disease, hyaline vascular variant
Follicles are more widely spaced and have concentric mantle zones. Follicles are depleted of germinal center B-cells, and there are prominent FDCs. Lymphoid cells of the atretic follicles are negative for BCL6 by IHC, and residual germinal center B-cells do not express BCL2 [23].
Nodular lymphocyte-predominant Hodgkin lymphoma
NLPHL has larger nodules than FL, and the nodules are more vaguely circumscribed. Most cells in nodules are small round lymphocytes. Centrocytes and centroblasts are absent. The presence of lymphocyte predominant (LP) cells which are negative for CD10 and BCL2, is the clue to the diagnosis of NLPHL. Interestingly, LP cells might express some germinal-center-associated markers, including BCL6, HGAL, LMO2, which may add to the difficulty of differentiating NLPHL B-cell rich nodular areas from FL [70]. In NLPHL, t(14;18)(q32;q21) is not identified [22].
Lymphocyte-rich classic Hodgkin lymphoma
There are commonly large nodules with eccentrically located germinal centers and expanded mantle zones in this neoplasm. There are Reed-Sternberg and Hodgkin cells, usually present within the expanded mantle zones. Reed-Sternberg and Hodgkin cells are positive for CD15, CD30, and PAX5 (dim) and negative for CD45, OCT2, BOB1, and CD79a. BCL6 positive germinal center B-cells do not co-express BCL2.
Mantle cell lymphoma
Mantle cell lymphoma (MCL) can resemble low-grade FL if it has in nodular or mantle zone pattern. Lymphoma cells of MCL are usually positive for CD5, CD43, cyclin D1, and negative for CD10.t(11;14)(q13;q32)/IGH-CCND1 is the hallmark of MCL.
Nodal marginal zone lymphoma
In nodal marginal zone lymphoma (MZL) lymphocytes with monocytoid features, frequent plasmacytic differentiation, and colonization of germinal centers by a monotypic and monoclonal B-cell population is identified. The neoplastic cells express pan-B-cell markers and are BCL2 positive by IHC. However, they are negative for CD5, CD10, cyclin D1, BCL6, LMO2, and other germinal center-associated markers. Expression of germinal center cell markers, including dual expression of BCL2 and BCL6 in rare cases of MZL, especially extranodal MZL, is an essential diagnostic pitfall to be aware of [71]. Positive IHC stain for IRTA1 and MNDA favor the diagnosis of MZL [72].
PROGNOSTIC FACTORS AND THERAPEUTIC MODALITIES
FL patients have a median overall survival of >15 years with different 5-year progression-free survival based on disease characteristics, comorbidities, therapies used, and therapeutic response [73]. Short remission after treatment has a poor prognosis [74].
FL may transform into an aggressive lymphoma. In ~25% to 35% of cases, FL progress to DLBCL, usually with the DLBCL showing biologic similarity to germinal center derived DLBCL [75-77]. As mentioned previously, a small subset of FL may progress to high-grade B-cell lymphomas with MYC, BCL2, and/or BCL6 translocations, and rare cases may progress to lymphoblastic lymphoma or relapse as classic Hodgkin lymphoma.
Other clinical prognostic factors in FL include:
Follicular Lymphoma International Prognostic Index (FLIPI), based on the following criteria, was published before the era of rituximab therapy [78]:
- Age (≥60 vs. <60 years)
- Ann Arbor stage (III–IV vs. I–II)
- Hemoglobin level (<120 vs. ≥120 g/L)
- Number of nodal areas involved (>4 vs. ≤4)
- Serum LDH level (above normal versus normal or below)
Three risk groups based on FLIPI are: low risk (score 0–1), intermediate-risk (score 2), and high risk (score 3–5), predicting 10-year overall survival of 70%, 50%, and 35%, respectively.
FLIPI2 system (in patients treated with rituximab) [79]:
- High serum β2 microglobulin
- Largest involved lymph node >6 cm
- BM involvement
- Hemoglobin <12 g/dL
- Age >60 years
M7 FLIPI better characterizes prognostic risk by incorporating the mutational status of seven genes included in this model: EZH2, ARID1A, MEF2B, EP300, FOXO1, CREBBP, and CARD11 [80].
Other histopathologic adverse prognostic factors include:
- High histologic grade (especially grade 3B) and presence of DLBCL [81]
- Low-grade FL or grade 3A FL with a high proliferation index (>30%) behaves similar to high-grade FL [54]
Cytogenetic adverse prognostic factors:
- Complex karyotype
- Both BCL2 and MYC rearrangement
TREATMENT
Standardized first-line therapy for FL [10]:
(1) Stage I or contiguous stage II (non-bulky): localized radiotherapy+/-systemic chemotherapy
(2) Stage I or stage II (bulky), or non-contiguous stage II: Anti-CD20 monoclonal antibody±chemotherapy; anti-CD20 monoclonal antibody±chemotherapy and/or radiotherapy
(3) Stage III or stage IV: anti-CD20 monoclonal antibody (in those with low tumor burden or elderly); anti-CD20 monoclonal antibody+chemotherapy (+/-maintenance anti-CD20)
Patients with advanced-stage disease and a low tumor burden also can undergo watchful waiting.
FL is generally very responsive to radiation and chemotherapy. Radiation alone can provide a long-lasting remission in some patients with limited disease. In more advanced stages, physicians may use one or more chemotherapy drugs or the monoclonal antibody rituximab (Rituxan) alone or with other agents.
Standard combination regimens include:
- R-Bendamustine (rituximab and bendamustine)
- R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone)
- R-CVP (rituximab, cyclophosphamide, vincristine, and prednisone)
- Chemotherapy, radiation, monoclonal antibodies, and chimeric antigen receptor T cell therapy, like axicabtagene ciloleucel, may be used to treat relapsed/refractory FL [84].
The bispecific antibody, mosunetuzumab, which targets CD20 and CD3, redirects and recruits endogenous T-cells to the proximity of CD20-expressing B-cells; it has promising clinical activity in patients with relapsed or refractory FL [85].
Standard second-line regimens include:
- Bendamustine (Treanda) with or without rituximab (Rituxan) or obinutuzumab (Gazyva) [86]
- Copanlisib (Aliqopa): phosphoinositide 3-kinase (PI3K) inhibitor [87]
- Duvelisib (Copiktra): PI3K inhibitor [88]
- Idelalisib (Zydelig): PI3K inhibitor [88]
- Umbralisib (UKONIQ): PI3K inhibitor [89]
- Tazemetostat (TAZVERIK): inhibitor of EZH2 [90]
- Abexinostat, vorinostat, and mocetinostat: Histone deacetylase inhibitors [91,92]
- Polatuzumab vedotin, an antibody–drug conjugate targeting the CD79b component of the BCR [93]
- Hu5F9-G4, an antibody targeting CD47 (which is overexpressed on cancer cells): enables the killing of tumor cells by macrophages by disrupting the inhibitory effect of CD47 on macrophage phagocytosis [94].
CONCLUSION
FL is the most common indolent B-cell lymphoma and originates from the lymphoid follicle’s germinal center B-cells (centrocytes and centroblasts).
In summary, we discussed the importance of morphologic classification and interpretation of ancillary studies in the accurate diagnosis of FL. Although many cases of FLs are typical and histopathologic features are straightforward, differentiating FL from mimickers, either from other lymphomas or reactive conditions, requires awareness of different morphologic patterns in FL and pitfalls in the interpretation of ancillary tests.
The overall survival for most patients is prolonged, but relapses are frequent. Understanding mutational abnormalities and signaling pathways, in addition to the t(14;18) (q32;q21) translocation and BCL2 mutation, will further help to identify innovative treatment approaches and application of immunotherapy and targeted therapy in patients with FL.
Notes
Ethics Statement
Not applicable.
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Writing—review & editing: MK, JRC. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest to disclose.
Funding Statement
No funding to declare.