Primary leiomyosarcoma of the bone: a case report
Article information

Abstract
Primary leiomyosarcoma of the bone is rare. Histologically, it resembles leiomyosarcoma of soft tissue. Given the rarity of this entity, its diagnosis should be made only after clinical studies and workup have excluded metastasis from other sites. Herein, we describe an additional case of primary bone leiomyosarcoma. We report a 32-year-old female patient, who presented with right knee pain and was found to have a right distal femur mass by imaging studies. Biopsy showed a neoplasm composed of fascicles of spindle cells, arranged in different patterns, with significant pleomorphism. The tumor cells were positive for smooth muscle actin, focally positive for desmin and H-caldesmon. No other masses in the body were detected by imaging studies. The diagnosis of leiomyosarcoma of the bone was rendered. Given the broad diagnostic differential of primary bone leiomyosarcoma, it is important to be aware of this rare bone tumor phenotype and of its histomorphologic and immunohistochemical features for an accurate diagnosis.
Primary bone leiomyosarcoma accounts for less than 0.7% of primary malignant bone neoplasms [1]. It has no known etiology, though a few cases have been associated with previous radiation therapy or Epstein-Barr virus (EBV) infection [2]. It most commonly arises from the distal femur [3]. Histologically, it resembles leiomyosarcoma of soft tissue. Immunohistochemical findings demonstrate diffuse positivity for smooth muscle actin (SMA), desmin, and H-caldesmon [2]. Given the rarity of primary bone leiomyosarcoma, its diagnosis should always be made only after thorough clinical studies and workup have excluded metastasis from other sites.
The main differential diagnoses of primary bone leiomyosarcoma include osteosarcoma, chondrosarcoma, myxofibrosarcoma, and synovial sarcoma among others [4]. Few cases of primary bone leiomyosarcoma have been reported in the literature [5-7], therefore, the information regarding its clinical and histological features, biological behavior, prognosis, and treatment modalities is still limited. Herein, we describe an additional case of primary bone leiomyosarcoma, to further increase our understanding of this rare tumor and to help avoid potential diagnostic pitfalls. Increasing awareness of this rare tumor is also important to improve prognosis and guide treatment.
CASE REPORT
A 32-year-old, medically-free female patient presented with right knee pain of 10-day duration. The pain worsened at night and with physical activities. Physical examination was unremarkable. X-ray revealed a right distal femur intraosseous metaphyseal central lytic lesion with cortical thinning and soft tissue extension. Magnetic resonance imaging showed a well-defined central destructive lesion, completely replacing the medullary cavity of the distal third of the right femoral shaft for a 10 cm span in craniocaudal dimension, reaching the anterior aspect of both femoral condyles and the posterior aspect of the intercondylar fossa with multiple areas of cortical bone destruction (Fig. 1). The patient underwent a core needle biopsy of the right distal femur lesion.
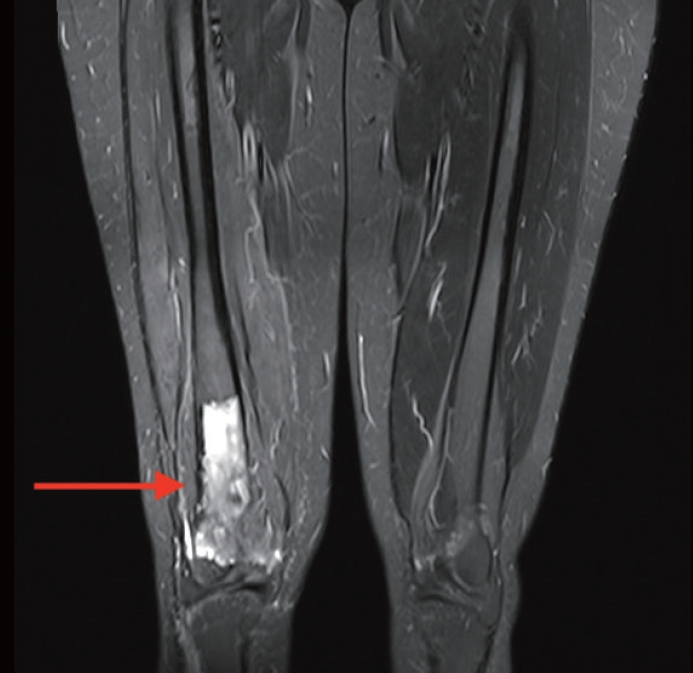
Magnetic resonance imaging shows an intraosseous metaphyseal central lytic lesion (red arrow), completely replacing the medullary cavity of the distal third of the right femoral shaft for a 10 cm span in craniocaudal dimension with multiple areas of cortical bone destruction.
Histopathologic findings
Gross examination revealed multiple tan-pink irregular friable focally hemorrhagic soft tissue fragments, measuring 2.5 × 1.7 × 0.4 cm in aggregate. Microscopic examination demonstrated fascicles of spindle and epithelioid cells with plump, blunt-ended nuclei, occasional subnuclear vacuoles, and moderate brightly eosinophilic cytoplasm. There was significant pleomorphism in the form of hyperchromasia, high nucleus-to-cytoplasmic ratio, and irregular nuclear contours, arranged in intersecting, storiform, and haphazard patterns (Fig. 2A, B). Other cells showed vesicular nuclei and prominent nucleoli. Multinucleated giant cells were also identified. The background was sclerotic. Fat lobules infiltrated by the atypical cells were also seen (Fig. 2C). Mitotic activity was 5 per 10 high-power fields.
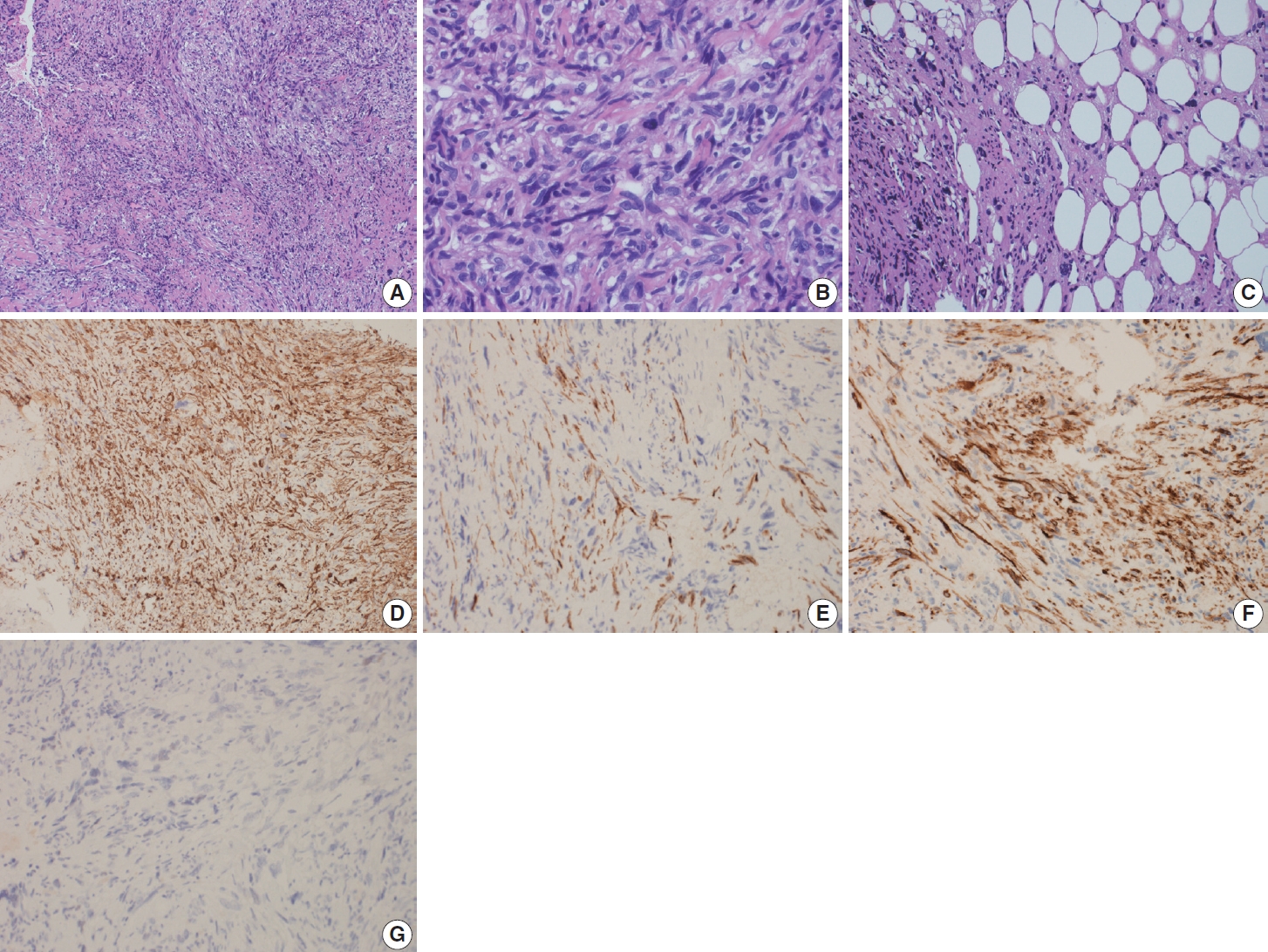
Microscopic and immunohistochemical features of primary bone leiomyosarcoma. (A) Photomicrograph shows fascicles of pleomorphic spindle and epithelioid cells arranged in fascicular, storiform, and haphazard patterns. (B) High-power view demonstrates cellular pleomorphism in the form of hyperchromasia, high nucleus-to-cytoplasmic ratios, and irregular nuclear contours. Some cells show vesicular nuclei and prominent nucleoli. (C) Photomicrograph shows fat lobules infiltrated by tumor cells. (D) The tumor cells are positive for smooth muscle actin. (E) The tumor cells show focal reactivity for Desmin. (F) The tumor cells show focal reactivity for H-caldesmon. (G) The tumor cells are negative for estrogen receptor.
Immunohistochemical findings
On immunohistochemical stains, performed by machine, the tumor cells were positive for SMA (ready-to-use, Dako, Glostrup, Denmark) “shown in Fig. 2D”, focally positive for desmin (readyto-use, Dako) (Fig. 2E), H-caldesmon (ready-to-use, Dako) (Fig. 2F), CD10 (ready-to-use, Dako), CD68 (ready-to-use, Dako) and SATB2 (prediluted, Cell Marque, Rocklin, CA, USA), and negative for cytokeratin (CK) AE1/AE3 (ready-to-use, Dako), CK MNF 116 (1:50, Dako), CDK4 (prediluted, Cell Marque), MDM2 (prediluted, Cell Marque), Melan-A (ready-to-use, Dako), S100 (ready-to-use, Dako), SOX-10 (prediluted, Cell Marque), estrogen receptor (ER; ready-to-use, Dako) (Fig. 2G), progesterone receptor (PR; ready-to-use, Dako), CD31 (ready-to-use, Dako), CD34 (ready-to-use, Dako), and myogenin (ready-to-use, Dako). S100 (ready-to-use, Dako) was positive in the adipocytes. Ki-67 (ready-to-use, Dako) proliferation index was high.
Final diagnosis and clinical course
The diagnosis of high-grade sarcoma, most consistent with leiomyosarcoma of the bone, was rendered. The patient had no previous gynecological history. Imaging studies revealed no uterine masses (Fig. 3) or soft tissue masses, and no lung or bony metastatic lesions. The patient underwent wide excision of the right distal femur. The gross examination of the resected specimen revealed an intramedullary irregular tan-white ill-defined infiltrative lesion measuring 9.2 × 6.3 × 3.2 cm. The final diagnosis on the excision specimen concurred with the biopsy diagnosis. The TNM stage was pT2 N0. The patient will be followed up by the oncology team on a regular basis.
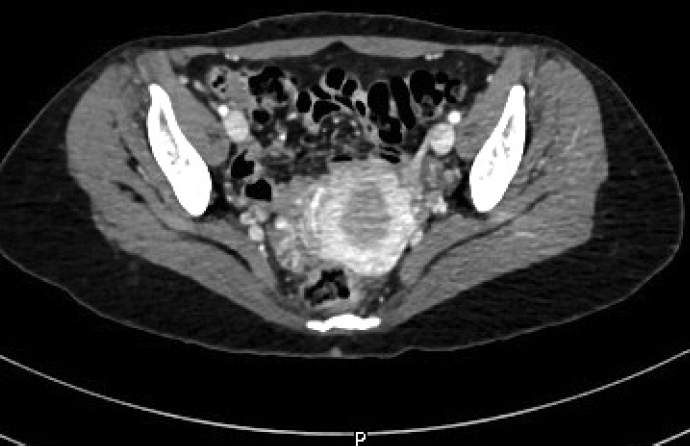
A computerized tomography scan of the pelvis reveals no uterine lesions.
DISCUSSION
Leiomyosarcomas are aggressive malignant soft tissue tumors arising from smooth muscle cells. They are common among soft tissue sarcomas, comprising approximately 25% of all sarcomas [8]. Primary bone leiomyosarcoma is a rare tumor, accounting for less than 0.7% of primary malignant bone neoplasms [1]. It was first described by Evans and Sanerkin in 1965 [5], and is slightly more common in males, with its incidence peaking at the fifth decade of life [2]. It mostly occurs at the distal femur followed by the proximal tibia and proximal humerus [3]. A small number of cases have been found to be associated with previous radiation therapy or EBV infection [2]. The most common symptom of primary bone leiomyosarcoma is pain, followed by the presence of a palpable mass and a pathologic fracture [9]. On radiographic examination, it usually presents as an intraosseous aggressive lytic lesion with a permeative pattern of osseous destruction [10]. The outline of low-grade lesions shows regular cortical destruction and is demarcated by a sclerotic rim whereas high-grade lesions have an ill-defined outline and lack a sclerotic rim. On gross examination, the lesions vary in size and have graytan creamy appearance with foci of necrosis and cystic degeneration [9]. Histologically, it resembles leiomyosarcoma of soft tissue with intersecting fascicles and bundles of pleomorphic spindle cells. The cells are characterized by eosinophilic cytoplasm and cigar-shaped nuclei with blunted ends. High-grade leiomyosarcomas are associated with marked pleomorphism, necrosis, and significant mitotic activity. Immunohistochemical findings demonstrate diffuse positivity for SMA, desmin, and H-caldesmon [2].
The cellular origin of primary bone leiomyosarcoma remains controversial. A vascular smooth muscle cell origin has been suggested due to the high vascularity of some of these lesions, while others have suggested that it can originate from multipotential mesenchymal stem cells capable of undergoing smooth muscle differentiation [11,12].
Given the rarity of primary bone leiomyosarcoma, metastasis from other sites should always be carefully excluded through clinical examination and imaging studies. In female patients, positivity for ER or PR immunostains hints toward a primary uterine origin [13]. In our case, the patient was medically free and had no previous history of leiomyosarcoma and her physical examination and pan–computed tomography findings were unremarkable. Based on this, the diagnosis of primary bone leiomyosarcoma was rendered.
In 1997, a case series on thirty-three patients with primary bone osteosarcoma was conducted [9]. In this study, they found that the patient’s age at diagnosis ranged from 13 to 77 years (average 44.4) with a male-to-female ratio of 1:1. The long bones were mostly affected, with distal femur and proximal tibia predominantly involved. Post-radiation bone leiomyosarcoma occurred in 15% of the patients, and seventy-five percent of the cases were high-grade with worse prognosis [9]. A recent case series was conducted on twenty-two patients with primary bone leiomyosarcoma. Researchers found that the patient’s age at diagnosis ranged from 17 to 73 years (average 45.5), and there was a slight male predominance. The lower limb was the most affected site. In this study, they reported 90.9% of the cases as high-grade, with distant metastasis reported in 40.9% of the patients and an overall survival rate of 59.1% in 5 years [14]. Compared to the previous studies, our case involves a 32-year-old female patient with no previous medical history or exposure to radiation. The tumor was located at the right distal femur. The patient has not developed local recurrence or metastasis since the time of diagnosis and treatment.
The main differential diagnoses of primary bone leiomyosarcoma include osteosarcoma, chondrosarcoma, myxofibrosarcoma, synovial sarcoma, malignant peripheral nerve sheath tumor (MPNST), fibrosarcoma, undifferentiated pleomorphic sarcoma and metastatic carcinoma [4]. The lack of malignant osteoid can help in excluding osteosarcoma, though this may be difficult in fibroblastic osteosarcomas since osteoid is focally present in such cases. Osteosarcomas may express desmin which will immunophenotypically overlap with leiomyosarcomas. Therefore, osteosarcomas should be carefully excluded after good radiological investigation and extensive sampling [4]. The lack of malignant cartilage production rules out chondrosarcoma. Synovial sarcoma usually demonstrates monotonous spindled cells with scant amphophilic cytoplasm, has a specific chromosomal translocation t(X;18) (p11;q11) and usually expresses TLE1, epithelial membrane antigen, and keratins. Features favoring leiomyosarcoma against MPNST include blunt-ended cigar-shaped nuclei with bright eosinophilic cytoplasm, positivity for SMA, H-caldesmon, and desmin. The diagnosis of fibrosarcoma and undifferentiated pleomorphic sarcoma is made by exclusion of other entities as they lack specific immunohistochemical and genetic markers [2,4]. Metastatic carcinomas express epithelial markers and lack myogenic markers. Lack of well-differentiated liposarcomatous component, MDM2 and CDK4 immunostains expression, and 12q13-15 amplification rules out dedifferentiated liposarcoma.
Data regarding the treatment modalities and prognosis of primary bone leiomyosarcoma is still limited due to its low prevalence. In one study, researchers reported that half of the patients with primary bone leiomyosarcoma developed metastases within one year of diagnosis [15].
In summary, we describe an additional case of primary bone leiomyosarcoma, which adds on more data about this rare category of bone tumors. In our case, the patient was medically free, had no previous history of leiomyosarcoma and no previous gynecological history, and had no uterine or soft tissue masses, along with the negative results of ER and PR in the tumor, which excludes the possibility of metastatic leiomyosarcoma from the uterus. Given the broad diagnostic differential of primary bone leiomyosarcoma, it is important to be aware of this rare bone tumor phenotype and its histomorphological and immunohistochemical features for accurate diagnosis.
Notes
Ethics Statement
The Institutional Review Board at Hamad Medical Corporation approved publication of this article under the number (MRC-04-22-670). Informed consent from the participant has been waived by Institutional Review Board.
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: SH. Data curation: AAD. Formal analysis: AAD. Investigation: AAD. Methodology: AAD. Supervision: SH. Validation: SH. Writing—original draft: AAD. Writing—review & editing: AAD, SH. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.