 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 49(5); 2015 > Article
-
Brief Case Report
Gastric Langerhans Cell Histiocytosis: Case Report and Review of the Literature - So Jung Lee1,2, Chung Su Hwang1,2, Gi Young Huh1,2, Chang Hun Lee1,2, Do Youn Park1,2
-
Journal of Pathology and Translational Medicine 2015;49(5):421-423.
DOI: https://doi.org/10.4132/jptm.2015.05.19
Published online: June 9, 2015
1Department of Pathology, Pusan National University Hospital, Pusan National University School of Medicine, Busan, Korea
2Biomedical Research Institute, Pusan National University Hospital, Busan, Korea
- Corresponding Author Do Youn Park, MD, PhD Department of Pathology, Pusan National University Hospital, Pusan National University School of Medicine, 179 Gudeok-ro, Seo-gu, Busan 49241, Korea Tel: +82-51-240-7422, Fax: +82-51-242-7422, E-mail: pdy220@pusan.ac.kr
• Received: February 3, 2015 • Revised: May 4, 2015 • Accepted: May 19, 2015
© 2015 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
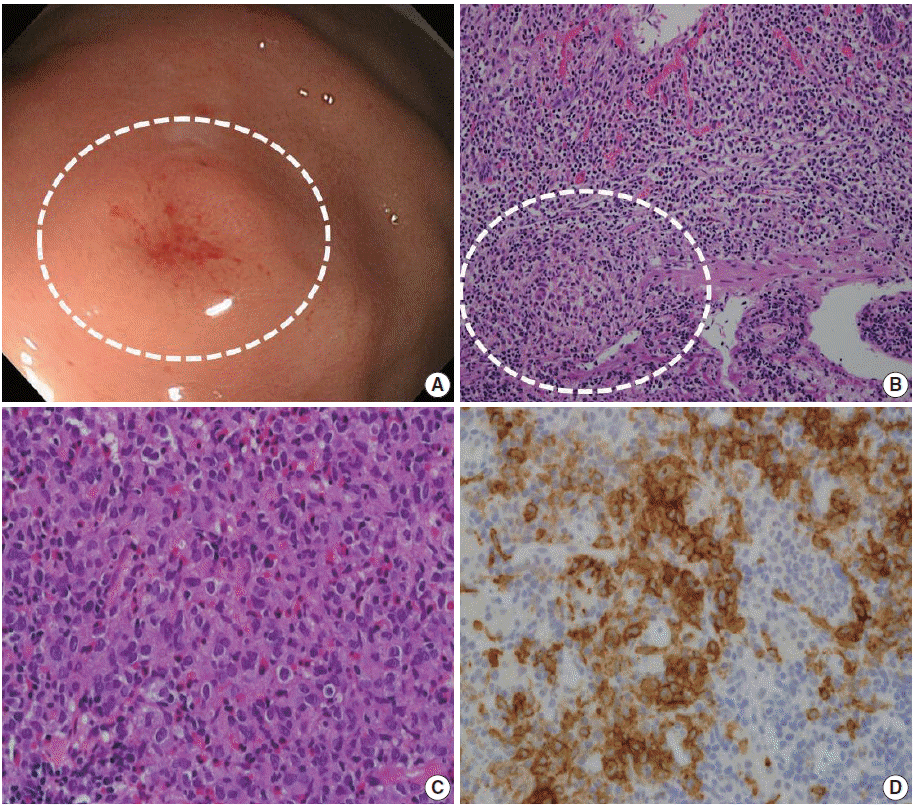
- A 64-year-old man was referred to a gastroenterologist for further evaluation of an abnormality found during an upper gastrointestinal examination. This patient had nothing remarkable in his medical history except medical treatment for hyperthyroidism. Esophagogastroduodenoscopy revealed an elevated mucosal lesion approximately 1 cm in size in the gastric fundus (Fig. 1A). The surface of the lesion was smooth with focal erosion. Microscopic examination of the endoscopic biopsy specimen revealed increased eosinophilia, and a cluster of histiocytic cells infiltrated the lamina propria and were mixed with lymphocytes, neutrophils, and plasma cells (Fig. 1C). The histiocytic cells had elongated nuclei, intranuclear grooves, and irregular nuclear membranes, as well as abundant fine granular eosinophilic to clear cytoplasm. Histiocytic cell clusters showed strong immunoreactivity to S100 and CD1a (Fig. 1D), while they were negative to cytokeratin AE1/AE3 and leukocyte common antigen. Through a combination of morphological and immunohistochemical analyses, a diagnosis of LCH was confirmed. A gastroenterologist performed ESD for complete removal of the lesion. The ESD specimen showed a very focal remnant LCH lesion that had been completely removed (Fig. 1B). Following complete resection of the gastric LCH lesion, a comprehensive evaluation was performed to determine the extent of the disease. No evidence of multisystem involvement was found. The patient’s 6-month follow-up visit revealed no local or systemic recurrence, and the patient remained in good health.
CASE REPORT
- LCH is a rare disease identified in both children and adults and is characterized by infiltration of histiocytic cells in various organs [3]. The pathogenesis and etiology of LCH are not fully understood [1]. A recent hypothesis suggests that LCH cells are derived from bone marrow monocyte precursors that differentiate into antigen-presenting cells (i.e., Langerhans cells or dendritic cells) in the epidermis, respiratory tract, and lymph nodes. The presence of monoclonality itself has been used as evidence that this disease is a clonal neoplastic disorder. Despite the neoplastic nature of the condition, the clinical course of LCH is heterogeneous. Therefore, treatment of LCH depends on the extent and severity of the disease at the time of diagnosis, which includes the number of involved organs and the presence of normal organ function [1].
- Involvement of the gastrointestinal tract is very rare and has been associated with systemic involvement and poor prognosis [4]. In pediatric patients, LCH in the gastrointestinal tract produces symptoms such as vomiting, loose stool, and abdominal pain and is related to a poor prognosis in the majority of neonatal cases; in these patients, skin lesions usually precede gastrointestinal LCH [4,5]. In contrast to children, adult patients are asymptomatic and present with solitary polypoids or elevated lesions [4]. Only six cases in adult of gastric LCH have been reported in the literature [4,6-10]. In contrast to pediatric LCH, adult gastric LCH appears as a unifocal disease without recurrence or progression, in a similar manner to our case (Table 1). Like many other gastric LCH cases, an elevated mucosal lesion was noted in our patient by the gastroenterologist, whose initial impression was a submucosal tumor. Endoscopic findings of gastric LCH mucosa are sometimes confused with gastric tumors presenting as polypoids and ulcerative mucosa [2,4]. Despite the absence of clinical features, gastric LCH shows the typical histological characteristics of LCH in other organs [3,4]. Histologically, histiocytic cells resemble Langerhans cells of the epidermis. Langerhans cell form a sheet of islands with indistinct cell borders and abundant eosinophilic cytoplasm. Their nuclei are elongated and irregular, exhibiting vesicular chromatin with a nuclear groove and often presenting with a single prominent nucleoli [3,4]. Langerhans cells are usually mixed with other inflammatory cells such as eosinophils, lymphocytes, plasma cells, and neutrophils. In the literature, predominant eosinophil infiltration has been reported in 50% of cases with gastric LCH [4]. In a similar manner to a previous report [4], our case showed aggregates of histiocytic cells with Langerhans cell nuclear features, which were mixed with inflammatory cells, mainly eosinophils. Immunohistochemical analysis is helpful to confirm the diagnosis, and Langerhans cells show diffuse immunoreactivity for S100 and CD1a [1,4]. In such cases, we suggest that stomach LCH could appear as a submucosal tumor upon endoscopy. Pathologists should consider the possibility of LCH if microscopic observation of biopsy specimens reveals increased eosinophilia or histiocytic cells. In these cases, immunohistochemical staining for S100 and CD1a is needed.
DISCUSSION
Acknowledgments
Fig. 1.Endoscopic and histologic finding of gastric Langerhans cell histiocytosis. (A) A mild elevated mucosal lesion (1 cm in size) with central erosion is observed upon gastroenteroscopy (circle). The lesion is located in the fundus of the stomach. (B) Microscopic analysis of the endoscopic submucosal dissection specimen. Focal histiocytic cell aggregates are present in the lamina propria and muscularis mucosa, with abundant eosinophils and other inflammatory cells. (C) Microscopic analysis of the endoscopic biopsy specimen reveals histiocytic cell aggregates in the lamina propria of the mucosa, with abundant eosinophil infiltration. Lymphocytes and plasma cells are also observed. The histiocytic cells show an irregular nuclear membrane and groove. These cells have abundant and granular eosinophilic to clear cytoplasm. (D) Immunohistochemistry for CD1a. The histiocytic cells show positive staining for CD1a.
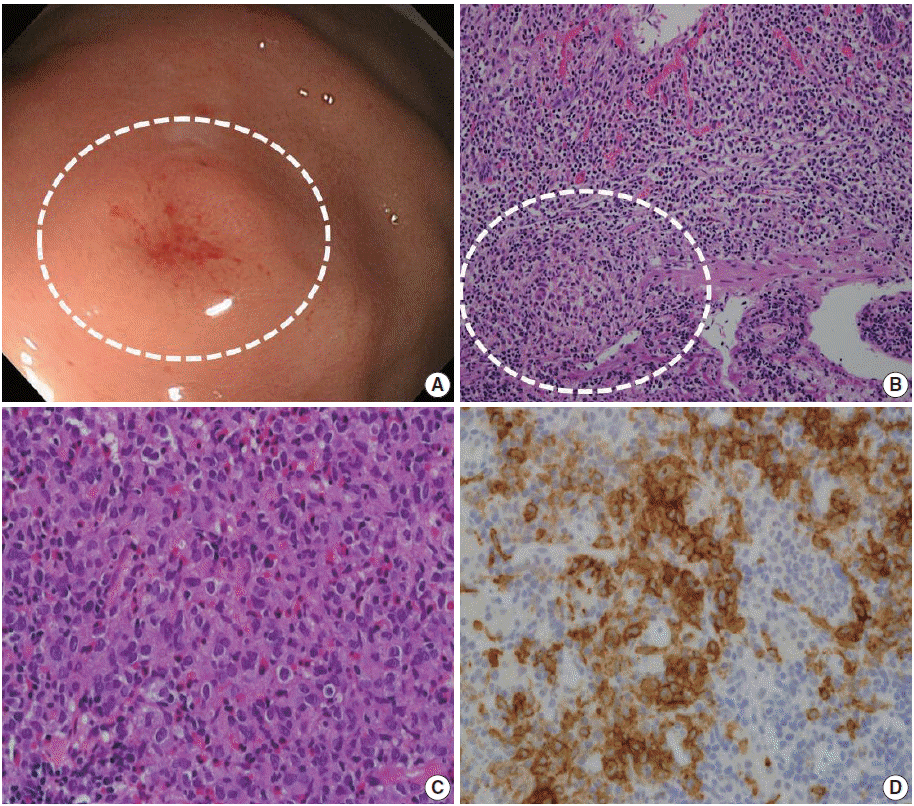
Table 1.Comparison of reported cases and presented case
| Case no. | Reference | Age (yr)/Sex | Symptom | Endoscopic finding | Site | Multiplicity | Specimen | Multisystema | Outcomes | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Singhi and Montgomery [4] | 68/M | Dysphasia | Polyp | Antrum | Solitary | Biopsy | Absent | Remission | 22 mo |
| 2 | Iwafuchi et al. [6] | 49/F | Asymptomatic | Sessile elevation | Throughout the stomach | Multiple | Biopsy | Absent | Remission | 5.6 yr |
| 3 | Nihei et al. [7] | 47/F | R/O cancer | Flat | Body | Solitary | Resection | Absent | Remission | 20 mo |
| 4 | Vazquez and Ayestaran [8] | 59/F | Epigastric pain | Ulcer | Lesser curvature | Solitary | Resection | N/A | N/A | N/A |
| 5 | Lee et al. [9] | 51/M | Asymptomatic | Elevated | Antrum | Solitary | Biopsy, ESD | Absent | Remission | 12 mo |
| 6 | Wada et al. [10] | 53/F | Abdominal discomfort | Polypoid | Throughout the stomach | Multiple | Biopsy | Absent | Alive/skin lesion developed | 2 yr |
| 7 | Present case | 64/M | Asymptomatic | Elevated | Fundus | Solitary | Biopsy, ESD | Absent | Remission | 6 mo |
- 1. Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: current concepts and treatments. Cancer Treat Rev 2010; 36: 354-9. ArticlePubMed
- 2. Behdad A, Owens SR. Langerhans cell histiocytosis involving the gastrointestinal tract. Arch Pathol Lab Med 2014; 138: 1350-2. ArticlePubMedPDF
- 3. Detlefsen S, Fagerberg CR, Ousager LB, et al. Histiocytic disorders of the gastrointestinal tract. Hum Pathol 2013; 44: 683-96. ArticlePubMed
- 4. Singhi AD, Montgomery EA. Gastrointestinal tract langerhans cell histiocytosis: a clinicopathologic study of 12 patients. Am J Surg Pathol 2011; 35: 305-10. PubMed
- 5. Vetter-Laracy S, Salinas JA, Martin-Santiago A, Guibelalde M, Balliu PR. Digestive tract symptoms in congenital langerhans cell histiocytosis: a fatal condition in an illness usually considered benign. J Pediatr Hematol Oncol 2014; 36: 426-9. PubMed
- 6. Iwafuchi M, Watanabe H, Shiratsuka M. Primary benign histiocytosis X of the stomach: a report of a case showing spontaneous remission after 5 1/2 years. Am J Surg Pathol 1990; 14: 489-96. PubMed
- 7. Nihei K, Terashima K, Aoyama K, Imai Y, Sato H. Benign histiocytosis X of stomach: previously undescribed lesion. Acta Pathol Jpn 1983; 33: 577-88. ArticlePubMed
- 8. Vazquez JJ, Ayestaran JR. Eosinophilic granuloma of the stomach similar to that of bone: light and electron microscopic study. Virchows Arch A Pathol Anat Histol 1975; 366: 107-11. PubMed
- 9. Lee CK, Lee SH, Cho HD. Localized Langerhans cell histiocytosis of the stomach treated by endoscopic submucosal dissection. Endoscopy 2011; 43 Suppl 2: E268-9. Article
- 10. Wada R, Yagihashi S, Konta R, Ueda T, Izumiyama T. Gastric polyposis caused by multifocal histiocytosis X. Gut 1992; 33: 994-6. ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Gastric Langerhans Cell Histiocytosis is Easily Ignored or Misdiagnosed in Stomach Biopsy and Indicates a Poor Outcome in Multisystem Langerhans Cell Histiocytosis
Yue Fan, Yang Liu, Haimin Xu, Lei Zhang, Fei Yuan, Hongmei Yi, Chaofu Wang
International Journal of Surgical Pathology.2026; 34(3): 640. CrossRef - Clinicopathological features of gastric Langerhans cell histiocytosis and a literature review
Xue Chen, Yunjin Wu, Xueli Wang, Muye Yang, Xiaoxiang Gao, Suxia Zhang, Huihui Sun, Yunhui Zhang, Hongmei Yi, Yu Zeng
Journal of Clinical and Experimental Hematopathology.2026; 66(1): 11. CrossRef - Solitary gastric langerhans cell histiocytosis treated by endoscopic submucosal dissection: A case report and literature review
Shengyue Zhou, Chunxiao Hu, Xiaohua Ye, Jin Ding, Hongjun Hua, Yilan Ma
Science Progress.2026;[Epub] CrossRef - Isolated Adult Gastrointestinal Tract Langerhans Cell Histiocytosis—Report of Two Rare Patients with Review of Literature
Ekta Jain, Eric Ollila, Fatme Ghandour, Abrar Alghamdi, Samuel Borak, Sameer Al Diffalha
International Journal of Surgical Pathology.2025; 33(7): 1679. CrossRef - Isolated Langerhans cell histiocytosis in the stomach of adults: four-case series and literature review
Jianmin Zhao, Yanlei Li, Yanlin Zhang, Xue Mei, Wei Liu, Yinghong Li
Journal of Hematopathology.2024; 17(2): 63. CrossRef - Clinical Characteristics and Outcomes in Patients With Localized Gastric Langerhans Cell Histiocytosis: A Case Series
Tae-Se Kim, Soomin Ahn, Yang Won Min, Hyuk Lee, Jun Haeng Lee, Poong-Lyul Rhee, Jae J. Kim, Byung-Hoon Min
The Korean Journal of Helicobacter and Upper Gastrointestinal Research.2024; 24(2): 175. CrossRef - Isolated Langerhans cell histiocytosis of the stomach in adults: An analysis of clinicopathologic characteristics and molecular genetics
Ruinuan Wu, Yali Zhao, Xikang Wu, Huihui Gui, Xia Liu, Zhaohui Liu
Medicine.2024; 103(51): e40950. CrossRef - Unifocal Gastric Langerhans Cell Histiocytosis in a Child—A Unique Case to Remember
Bhaswati C. Acharyya, Mandira Roy, Hema Chakraborty
JPGN Reports.2022;[Epub] CrossRef - Langerhans Cell Histiocytosis with the Synchronous Invasion of Stomach and Colon in an Adult Patient: A Case Report
Seong Je Kim, Se In Hah, Ji Yoon Kwak, Jung Woo Choi, Hyun Chin Cho, Chang Yoon Ha, Woon Tae Jung, Ok Jae Lee, Chang Min Lee
The Korean Journal of Gastroenterology.2022; 80(3): 149. CrossRef - Gastrointestinal Langerhans cell histiocytosis with unifocal, single‐system involvement in adults: Cases report and literature review
Li Wang, Fang Yang, Yong Ding, Lixia Lu, Haili Li, Yangyang Cui, Lu Lu, Xiaohan Shen, Rong Ge
Journal of Clinical Laboratory Analysis.2022;[Epub] CrossRef - Upper Gastrointestinal Langerhans Cell Histiocytosis: A Report of 2 Adult Cases and a Literature Review
Yui Matsuoka, Yoshiki Iemura, Masakazu Fujimoto, Shinsuke Shibuya, Atsushi Yamada, Shigehiko Fujii, Toshihiro Kusaka, Takero Shindo, Sachiko Minamiguchi, Hironori Haga
International Journal of Surgical Pathology.2021; 29(5): 550. CrossRef - Langerhans cell histiocytosis of the gastrointestinal tract
Aoife J. McCarthy, Madiha Emran Soofi, Imaad Mujeeb, Runjan Chetty
Diagnostic Histopathology.2018; 24(4): 154. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
Gastric Langerhans Cell Histiocytosis: Case Report and Review of the Literature

Fig. 1. Endoscopic and histologic finding of gastric Langerhans cell histiocytosis. (A) A mild elevated mucosal lesion (1 cm in size) with central erosion is observed upon gastroenteroscopy (circle). The lesion is located in the fundus of the stomach. (B) Microscopic analysis of the endoscopic submucosal dissection specimen. Focal histiocytic cell aggregates are present in the lamina propria and muscularis mucosa, with abundant eosinophils and other inflammatory cells. (C) Microscopic analysis of the endoscopic biopsy specimen reveals histiocytic cell aggregates in the lamina propria of the mucosa, with abundant eosinophil infiltration. Lymphocytes and plasma cells are also observed. The histiocytic cells show an irregular nuclear membrane and groove. These cells have abundant and granular eosinophilic to clear cytoplasm. (D) Immunohistochemistry for CD1a. The histiocytic cells show positive staining for CD1a.
Fig. 1.
Gastric Langerhans Cell Histiocytosis: Case Report and Review of the Literature
| Case no. | Reference | Age (yr)/Sex | Symptom | Endoscopic finding | Site | Multiplicity | Specimen | Multisystem |
Outcomes | Follow-up |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Singhi and Montgomery [4] | 68/M | Dysphasia | Polyp | Antrum | Solitary | Biopsy | Absent | Remission | 22 mo |
| 2 | Iwafuchi et al. [6] | 49/F | Asymptomatic | Sessile elevation | Throughout the stomach | Multiple | Biopsy | Absent | Remission | 5.6 yr |
| 3 | Nihei et al. [7] | 47/F | R/O cancer | Flat | Body | Solitary | Resection | Absent | Remission | 20 mo |
| 4 | Vazquez and Ayestaran [8] | 59/F | Epigastric pain | Ulcer | Lesser curvature | Solitary | Resection | N/A | N/A | N/A |
| 5 | Lee et al. [9] | 51/M | Asymptomatic | Elevated | Antrum | Solitary | Biopsy, ESD | Absent | Remission | 12 mo |
| 6 | Wada et al. [10] | 53/F | Abdominal discomfort | Polypoid | Throughout the stomach | Multiple | Biopsy | Absent | Alive/skin lesion developed | 2 yr |
| 7 | Present case | 64/M | Asymptomatic | Elevated | Fundus | Solitary | Biopsy, ESD | Absent | Remission | 6 mo |
Table 1. Comparison of reported cases and presented case
M, male; F, female; R/O, rule out; N/A, not applicable; ESD, endoscopic submucosal dissection. Multisystem involvement at diagnosis.

