 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 50(3); 2016 > Article
-
Original Article
Non-small Cell Lung Cancer with Concomitant EGFR, KRAS, and ALK Mutation: Clinicopathologic Features of 12 Cases - Taebum Lee, Boram Lee, Yoon-La Choi, Joungho Han, Myung-Ju Ahn1, Sang-Won Um2
-
Journal of Pathology and Translational Medicine 2016;50(3):197-203.
DOI: https://doi.org/10.4132/jptm.2016.03.09
Published online: April 18, 2016
Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
1Division of Hematology-Oncology, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
2Division of Pulmonary and Critical Care Medicine, Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
- Corresponding Author: Joungho Han, MD Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 06351, Korea Tel: +82-2-3410-2800 Fax: +82-2-3410-0025 E-mail: hanjho@skku.edu
© 2016 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background:
- Although epidermal growth factor receptor (EGFR), v-Ki-ras2 Kirsten rat sarcoma viral oncogene (KRAS), and anaplastic lymphoma kinase (ALK) mutations in non-small cell lung cancer (NSCLC) were thought to be mutually exclusive, some tumors harbor concomitant mutations. Discovering a driver mutation on the basis of morphologic features and therapeutic responses with mutation analysis can be used to understand pathogenesis and predict resistance in targeted therapy.
-
Methods:
- In 6,637 patients with NSCLC, 12 patients who had concomitant mutations were selected and clinicopathologic features were reviewed. Clinical characteristics included sex, age, smoking history, previous treatment, and targeted therapy with response and disease-free survival. Histologic features included dominant patterns, nuclear and cytoplasmic features.
-
Results:
- All patients were diagnosed with adenocarcinoma and had an EGFR mutation. Six patients had concomitant KRAS mutations and the other six had ALK mutations. Five of six EGFR-KRAS mutation patients showed papillary and acinar histologic patterns with hobnail cells. Three of six received EGFR tyrosine kinase inhibitor (TKI) and showed partial response for 7–29 months. All six EGFR-ALK mutation patients showed solid or cribriform patterns and three had signet ring cells. Five of six EGFR-ALK mutation patients received EGFR TKI and/or ALK inhibitor and four showed partial response or stable disease, except for one patient who had acquired an EGFR mutation.
-
Conclusions:
- EGFR and ALK mutations play an important role as driver mutations in double mutated NSCLC, and morphologic analysis can be used to predict treatment response.
- A retrospective review was performed of biopsied and/or surgically resected cases diagnosed as NSCLC and tested for EGFR, KRAS, and ALK mutations in the Department of Pathology and Translational Genomics at Samsung Medical Center, Seoul, Korea from 2006 to 2014. Of 6,637 NSCLC patients, 6,595 EGFR, 5,177 KRAS, and 4,869 ALK mutation tests were performed. Among them, 2,387 patients (36.2%) had EGFR, 398 (7.7%) had KRAS, and 281 (5.8%) had ALK mutations. Based on these tests, we selected 12 patients with concomitant mutations in the same tumor.
- Clinical data, including gender, age, smoking history, and previous concurrent chemoradiation therapy history were extracted from electronic medical records. All hematoxylin and eosin stained slides of selected cases were reviewed by two pathologists (T.L. and B.L.). Histologic classification was made according to the IASLC/American Thoracic Society (ATS)/European Respiratory Society (ERS) International Multidisciplinary Classification of Lung Adenocarcinoma, and typical pathologic features were re-evaluated, including cell type, nuclear and cytoplasmic characteristics.
- We used two methods to evaluate EGFR mutations in the 18th, 19th, 20th, and 21st exon: Sanger sequencing and real time polymerase chain reaction after peptide nucleic acid (PNA)–clamping using the PNA clamping EGFR Mutation Detection Kit (Panagene, Inc., Daejeon, Korea). KRAS mutations were evaluated with Sanger sequencing in the 12th and 13th exon. Extracted genomic DNA isolated from formalin-fixed paraffin-embedded (FFPE) tissue was used for EGFR and KRAS analyses. ALK mutation tests were performed using immunohistochemical (IHC) (1:40, NCL-ALK, clone 5A4, Novocastra, Newcastle upon Tyne, UK) and fluorescence in situ hybridization (FISH) analysis (LSI ALK dual color break-apart probe, Dako, Glostrup, Denmark) with FFPE tissue. Diffuse strong positivity in the cytoplasm was regarded as positive in ALK IHC. In the FISH analysis, 50 non-overlapping nuclei were counted and 15% of break-apart probes were used as a cutoff value for positivity. Diffuse strong positive ALK IHC results were interpreted as a surrogate marker of ALK rearrangement [14].
MATERIALS AND METHODS
- Clinical characteristics
- Clinical data and pathologic features of the 12 patients with concomitant mutations are summarized in Table 1. The age at diagnosis of patients ranged from 48 to 78 years old (median, 62 years old) and 75% of patients were female. All three male patients had a history of smoking (15–57 pack-years) in contrast to none of female patients. Five surgically resectable patients underwent lobectomy and seven clinical stage IV patients had a needle aspiration or biopsy from the lung, bronchus, lymph node, or adrenal gland.
- Patient No. 5 received EGFR TKI targeted therapy for bone metastasis at 15 months after lobectomy. Patient No. 6 refused treatment with chemo- or targeted therapy and patient No. 7 had a history of lobectomy at an outside hospital without any other treatment 9 years prior to the lymph node biopsy. Patient No. 10 received neoadjuvant concurrent chemoradiation therapy prior to surgery. Patient No. 12 previously underwent lobectomy and was treated with gefitinib for 1 month and crizotinib for 4 months in China. She had no EGFR or KRAS mutation at the time of diagnosis at China. Aside from this patient, all other patients had no history of prior targeted therapy before mutation testing.
- Mutational and histologic characteristics
- All 12 patients were diagnosed with adenocarcinoma and had an EGFR mutation. EGFR mutations of three patients were only detected with the PNA clamping method, while nine were confirmed by Sanger sequencing. Five (41.7%) had the missense L858R mutation in exon 21, four (33.3%) had the missense G719X mutation in exon 18, and three (25%) had a deletion in exon 19. Patient No. 8 had the L858R and G719X mutations at the same time and patient No. 12 had a missense R803W mutation at exon 20 that was not identified before targeted therapy at the outside hospital. No patient had a T790M point mutation, which is associated with resistance. Six patients had additional KRAS mutations and the other six had an ALK mutation (referred to as ‘EGFR-KRAS’ and ‘EGFR-ALK’ hereafter). Three among the six patients who showed positivity at ALK IHC were confirmed with FISH analysis.
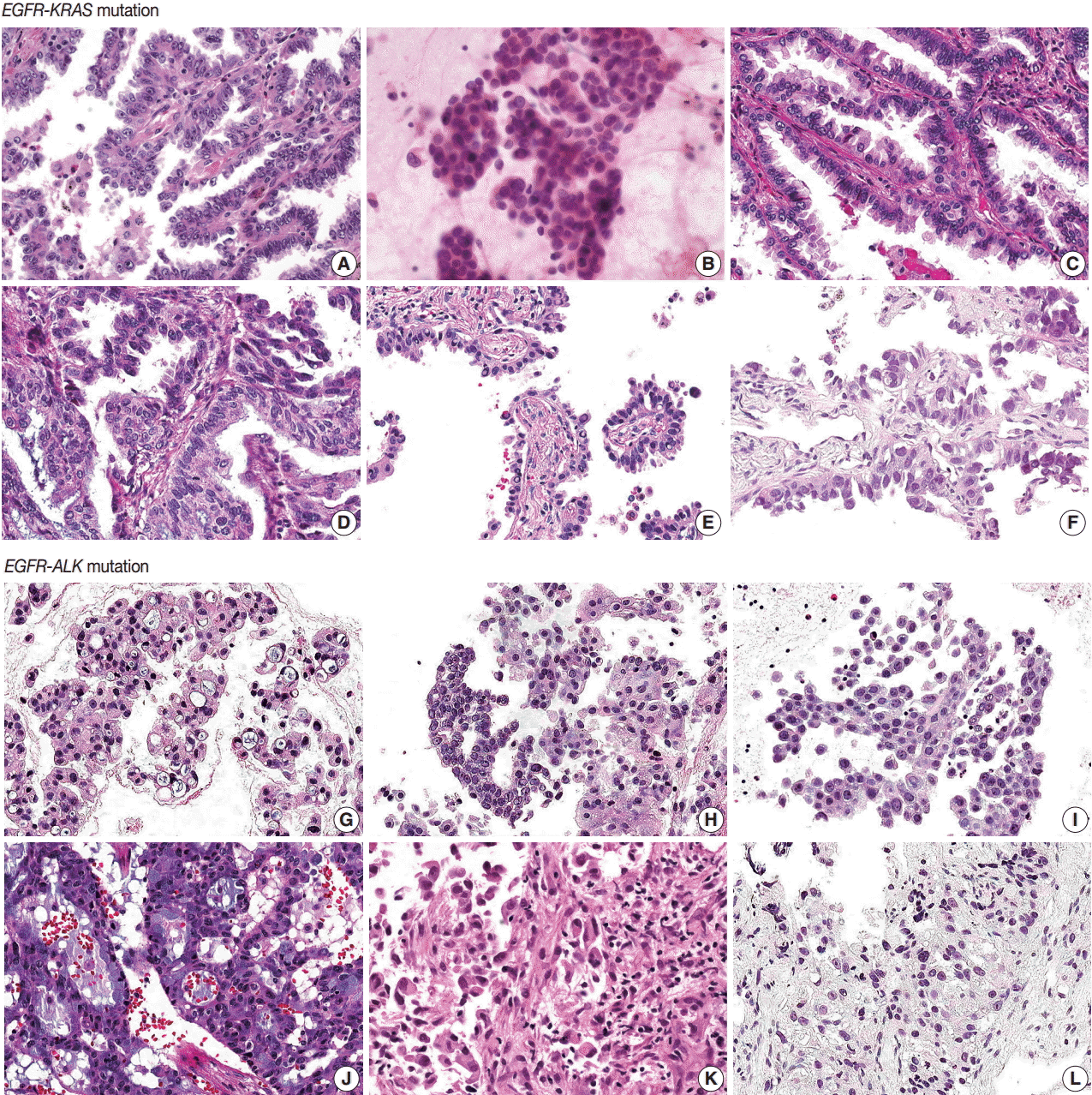
- EGFR-KRAS and EGFR-ALK tumors showed distinct morphologic characteristics. Six EGFR-KRAS patients had papillary, acinar, solid, and micropapillary patterns (Fig. 1). Among them, five patients had typical hobnail cell features with apical snouts; the remaining patient (patient No. 2) underwent needle aspiration and did not show any typical cell features. One patient (patient No. 4) had a focal columnar cell component and intraand extracytoplasmic mucin, which are similar morphologic features of NSCLC with a KRAS mutation. The other five patients did not show any mucin. Two of the six patients had intranuclear inclusions.
- In contrast, six EGFR-ALK patients showed solid, cribriform and micropapillary patterns. Three of six patients had signet ring cells and intracytoplasmic mucin with or without extracytoplasmic mucin production, which are typical features of ALK-positive NSCLC. The others did not show any typical cell features or mucins. One patient had intranuclear inclusions.
- Treatment responses and follow-up status
- Seven patients had surgical resection and two were alive without targeted therapy and any evidence of relapse or progression, including one patient who received neoadjuvant concurrent chemoradiation therapy. One patient who had surgery and decided not to receive targeted therapy due to old age and underlying diabetes mellitus died of disease after 53 months from surgery (patient No. 1).
- EGFR TKI included gefitinib (250 mg, orally, every day) and erlotinib (150 mg, orally, every day) and the ALK inhibitor was crizotinib (250 mg, orally, every day or twice daily). Three among six EGFR-KRAS patients received targeted therapy with EGFR TKI. All three patients showed partial responses, but two also showed disease progression after 7 and 20 months. In the EGFR-ALK patients, two were treated with EGFR TKI, two with ALK inhibitor and one received both EGFR TKI and ALK inhibitor therapy. Most patients treated with ALK inhibitor or EGFR TKI showed partial response or stable disease, but patient No. 12 with EGFR TKI showed progressive disease.
RESULTS
- Concomitant genetic alteration of NSCLC is unusual because EGFR, KRAS, and ALK mutations are widely known as mutually exclusive. Most are associated with acquired mutation after targeted therapy and are related to drug resistance. In previous studies, two different hypotheses were proposed to explain dual mutation in NSCLC [12]. Underlying intratumoral heterogeneity of EGFR mutations [15], different tumor cells may have different oncogenic driver mutations. Some authors have also suggested coexistence of mutations in the same tumor cells using IHC expression and mutation-specific antibodies [12,16].
- EGFR, as a growth factor membrane-bound receptor tyrosine kinase, controls cell proliferation and survival. Approximately 36% of patients with NSCLC in East Asia have EGFR mutations and 90% of mutations are exon 21 L858R or exon 19 deletion [17]. These mutations increase tyrosine kinase activity, resulting in hyperactivation of the RAS-RAF-MEK-ERK pathway, which regulates cell proliferation, and the phosphoinositide 3-kinase–AKT–mammalian target of rapamycin pathway, which is associated with cell survival. RAS proteins are central mediators in EGFR tyrosine kinase signaling and also associated with cell proliferation and differentiation. In particular, KRAS mutations are common in lung cancer but less common in East Asia than in Western countries [18]. ALK is a receptor tyrosine kinase. ALK fusion with various N-terminal fusion partners stimulates kinase activity, and signaling of these mutations are involved in cell growth, proliferation and apoptosis. Among NSCLCs, 3%–7% harbor ALK fusion [3,4] and the majority of these fusions are associated with the echinoderm microtubule-associated proteinlike 4 (EML4) gene. EML4-ALK fusions are associated with EGFR TKI resistance [19].
- Typical histologic features of lung cancer with EGFR, KRAS, and ALK mutations have been described [20-24]. All of these mutations are frequent in adenocarcinoma and EGFR mutations are more common in tumors with papillary, micropapillary, acinar, and lepidic patterns than in others. Hobnail cell type and intranuclear inclusion are typical nuclear features of EGFR mutation-harboring tumors. A solid pattern and mucinous type are more frequent in KRAS mutation tumors. Extracellular mucin production, cribriform patterns, and signet ring cell features are more common in ALK mutation tumors.
- In this study, we analyzed six EGFR-KRAS (1.5%) and six EGFR-ALK patients (2.1%) among KRAS and ALK mutation-positive NSCLC patients. The majority of EGFR-KRAS tumors showed typical histologic features of EGFR mutation, except one case which had focal morphologic features similar to KRAS mutation. In a previous study, NSCLC harboring a KRAS mutation showed resistance to EGFR TKI and adjuvant chemotherapy [2]. In our study, however, three patients showed partial response to gefitinib and erlotinib for 7–29 months. Intratumoral heterogeneity of the mutation may explain this discordance between coexistent KRAS mutation and response to EGFR TKI therapy. Patient No. 2 had short progression-free survival (PFS) and disease progressed rapidly, while the other two patients had 20–29 months of PFS. Based on the evidence of morphologic phenotypes and responsiveness to targeted therapy, we expected these two patients to have coexistent mutations in the same tumor cells, and hypothesized that the EGFR mutation was the dominant driver oncogene in lung cancer, even if the tumor cells harbored an additional KRAS mutation.
- In EGFR-ALK patients, some previous studies have shown acquired EGFR mutations as a mechanism of resistance to ALK inhibitor therapy [25,26]. In our case, patient No. 12 had no EGFR or KRAS mutation and was treated with crizotinib in an outside hospital. After 4 months of ALK-targeted therapy, she acquired an additional EGFR mutation and showed poor response to erlotinib, which is consistent with previous studies. In the other four patients, however, diverse responses were identified with no resistance to gefitinib or crizotinib. Conflicting results of responsiveness to EGFR TKI and ALK inhibitor in EGFR-ALK patients in NSCLC have been reported in a few limited studies [10,27]. Recently, Yang et al. [16] investigated relationships between receptor phosphorylation and response to targeted therapy, and suggested that relative phospho-EGFR and ALK levels can be used to predict response.
- Although the G719X mutation accounts for about 7% of EGFR mutated NSCLC in East Asia [17], three of five EGFR-ALK patients had the EGFR exon 18 G719X missense mutation in our study. In addition, three of five EGFR-ALK patients showed typical histologic features of ALK-expressing adenocarcinoma. From these findings, we suspect that the ALK mutation determines morphological phenotypes and acts as a driver oncogene, and the EGFR mutation may also play an important role in oncogenesis. Accordingly, based on evaluation of the signaling pathway, including phosphorylation of receptor kinase, the modification or combination of EGFR TKI and ALK inhibitor offer promising treatments.
- The small number of cases limited our analysis. Baldi et al. [12] revealed the possibility of more frequent concomitant mutations than expected and Won et al. [13] reported an increased proportion of NSCLC harboring concomitant EGFR and ALK mutations from 4.4% to 15.4% using more sensitive methods such as targeted next-generation sequencing (NGS) and mutant-enriched NGS. Greater numbers of previously undisclosed concomitantly mutated tumors can be identified using more sensitive and advanced techniques. Some authors also suggest that tumor burden of each mutation affects responses to targeted therapy. There may be a limitation to evaluating mutations from biopsy specimens, but the majority of patients who need to receive targeted therapy might be clinically unresectable and may only be diagnosed with biopsy. Thus, the expectation of tumor burden in a limited biopsy specimen is unfavorable.
- To the best of our knowledge, this is the first study to describe double mutated NSCLC with histologic findings. Morphologic findings might help predict underlying driver mutations and further studies are warranted.
- In conclusion, we investigated genetic driver mutations in 12 double mutated NSCLCs with analysis of clinical and pathologic features. The majority of EGFR-KRAS tumors showed typical histologic patterns and cell features of EGFR-mutated tumors and partially responded to EGFR TKI. In EGFR-ALK tumors, however, some showed ALK-mutated features and partially responded to ALK inhibitor or EGFR TKI. EGFR and ALK play an important role in the oncogenesis of NSCLC and tumor morphology can provide important clues to predict treatment response. We suggest that patients with histologic features of EGFR mutations can be treated with EGFR TKI, even with coexistence of a KRAS mutation. We also suggest that EGFR-ALK patients should have underlying mutational pathways evaluated and be treated with a selection and/or combination of ALK inhibitor and EGFR TKI. Further studies are needed to support these interesting findings.
DISCUSSION
Acknowledgments
| Patient No. | Sex/Age(yr) | Smoking | Specimen | Stage | EGFR mutation | Additional mutation | Dominant pattern | Typical cell feature | Mucin | Intranuclear inclusion | Surgery | Targeted therapy | Response | Status | PFS (mo) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/74 | 57PY | Lung/R | pT1bN1 | E21 L858R | KRAS I21S | Papillary and micropapillary | Hobnail cells | No | No | + | - | - | DODa | - |
| 2 | F/62 | Never | Lung/A | M1(IV) | E21 L858R | KRAS G12D | b | No | No | Present | Gefitinib | PR → PD | DOD | 7 | |
| 3 | F/63 | Never | Lung/R | pT1bNO | E21 L858R | KRAS G13A | Papillary and acinar | Hobnail cells | No | No | + | - | - | NED | - |
| 4 | F/48 | Never | Lung/R | pT1bN2 | E19 deletion | KRAS G12V | Acinar and solid | Hobnail and columnar cells | Intra- and extracytoplasmic | No | + | Erlotinib | PR → PD | DOD | 20 |
| 5 | M/55 | 20PY | Lung/R | pT2N2 | E19 deletion | KRAS G13C | Solid and acinar | Hobnail cells | No | Present | + | Gefitinib | PR | AWD | 29 |
| 6 | F/78 | Never | Lung/B | M1(IV) | E18 G719X | KRAS G12D | Papillary and acinar | Hobnail cells | No | No | - | (refused) | - | AWDa | - |
| 7 | F/62 | Never | LN/B | M1(IV) | E21 L858Rc | ALK | Solid and cribriform | Signet ring cells | Intracytoplasmic | No | +d | Gefitinib | PR | AWDa | 18e |
| 8 | F/62 | Never | Lung/B | M1(IV) | E21 L858R | ALK | Solid and cribriform | No | No | No | - | Gefitinib and Crizotinib | SD and PR | AWD | 24 |
| E18 G719X | |||||||||||||||
| 9 | M/56 | 15PY | Lung/B | M1(IV) | E18 G719Xc | ALK | Solid | No | No | No | Crizotinib | SD | DOD | 4 | |
| 10 | F/68 | Never | Lung/R | ypT2N2 | E18 G719Xc | ALK | Solid, micropapillary and cribriform | Signet ring cells | Intra- and extracytoplasmic | Present | + | - | - | NED | - |
| 11 | F/58 | Never | Lung/B | M1(IV) | E19 deletion | ALK | Solid | Signet ring cells | Intracytoplasmic | No | - | Crizotinib | - | AWDa | e |
| 12 | F/66 | Never | Adrenal/B | M1(IV) | E20 R803W | ALK | Solid | No | No | No | +d | Erlotinib | PD | AWDa | 0.7 |
EGFR, epidermal growth factor receptor; PFS, progression-free survival; M, male; PY, pack-year; R, resection; E, exon; KRAS, v-Ki-ras2 Kirsten rat sarcoma viral oncogene; DOD, died of disease; F, female; A, aspiration; PR, partial response; PD, progressive disease; NED, no evidence of disease; AWD, alive with disease; B, biopsy; LN, lymph node; ALK, anaplastic lymphoma kinase; SD, stable disease.
aLost to follow-up;
bCannot identify dominant architectural patterns in an aspiration slide;
cMutation detected only in PNA clamping method;
dHave a history of lobectomy at outside hospital, prior to biopsy of metastatic lesion;
eCannot evaluate due to lost to follow-up.
- 1. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350: 2129-39. PubMed
- 2. Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc 2009; 6: 201-5. ArticlePubMed
- 3. Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363: 1693-703. PubMedPMC
- 4. Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448: 561-6. ArticlePubMedPDF
- 5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65: 5-29. ArticlePubMed
- 6. Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497-500. ArticlePubMed
- 7. Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol 2013; 8: 823-59. ArticlePubMedPMC
- 8. Narita Y, Matsushima Y, Shiroiwa T, et al. Cost-effectiveness analysis of EGFR mutation testing and gefitinib as first-line therapy for non-small cell lung cancer. Lung Cancer 2015; 90: 71-7. ArticlePubMed
- 9. Gainor JF, Varghese AM, Ou SH, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res 2013; 19: 4273-81. PubMed
- 10. Tiseo M, Gelsomino F, Boggiani D, et al. EGFR and EML4-ALK gene mutations in NSCLC: a case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer 2011; 71: 241-3. ArticlePubMed
- 11. Lee JK, Kim TM, Koh Y, et al. Differential sensitivities to tyrosine kinase inhibitors in NSCLC harboring EGFR mutation and ALK translocation. Lung Cancer 2012; 77: 460-3. ArticlePubMed
- 12. Baldi L, Mengoli MC, Bisagni A, Banzi MC, Boni C, Rossi G. Concomitant EGFR mutation and ALK rearrangement in lung adenocarcinoma is more frequent than expected: report of a case and review of the literature with demonstration of genes alteration into the same tumor cells. Lung Cancer 2014; 86: 291-5. ArticlePubMed
- 13. Won JK, Keam B, Koh J, et al. Concomitant ALK translocation and EGFR mutation in lung cancer: a comparison of direct sequencing and sensitive assays and the impact on responsiveness to tyrosine kinase inhibitor. Ann Oncol 2015; 26: 348-54. ArticlePubMed
- 14. Paik JH, Choe G, Kim H, et al. Screening of anaplastic lymphoma kinase rearrangement by immunohistochemistry in non-small cell lung cancer: correlation with fluorescence in situ hybridization. J Thorac Oncol 2011; 6: 466-72. ArticlePubMed
- 15. Bai H, Wang Z, Wang Y, et al. Detection and clinical significance of intratumoral EGFR mutational heterogeneity in Chinese patients with advanced non-small cell lung cancer. PLoS One 2013; 8: e54170. ArticlePubMedPMC
- 16. Yang JJ, Zhang XC, Su J, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res 2014; 20: 1383-92. ArticlePubMedPDF
- 17. Choi YL, Sun JM, Cho J, et al. EGFR mutation testing in patients with advanced non-small cell lung cancer: a comprehensive evaluation of real-world practice in an East Asian tertiary hospital. PLoS One 2013; 8: e56011. ArticlePubMedPMC
- 18. Sun Y, Ren Y, Fang Z, et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol 2010; 28: 4616-20. ArticlePubMedPMC
- 19. Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol 2009; 27: 4247-53. PubMedPMC
- 20. Ninomiya H, Hiramatsu M, Inamura K, et al. Correlation between morphology and EGFR mutations in lung adenocarcinomas significance of the micropapillary pattern and the hobnail cell type. Lung Cancer 2009; 63: 235-40. ArticlePubMed
- 21. Rekhtman N, Ang DC, Riely GJ, Ladanyi M, Moreira AL. KRAS mutations are associated with solid growth pattern and tumor-infiltrating leukocytes in lung adenocarcinoma. Mod Pathol 2013; 26: 1307-19. ArticlePubMedPMCPDF
- 22. Nishino M, Klepeis VE, Yeap BY, et al. Histologic and cytomorphologic features of ALK-rearranged lung adenocarcinomas. Mod Pathol 2012; 25: 1462-72. ArticlePubMedPDF
- 23. Ha SY, Choi SJ, Cho JH, et al. Lung cancer in never-smoker Asian females is driven by oncogenic mutations, most often involving EGFR. Oncotarget 2015; 6: 5465-74. ArticlePubMedPMC
- 24. Choi IH, Kim DW, Ha SY, Choi YL, Lee HJ, Han J. Analysis of histologic features suspecting anaplastic lymphoma kinase (ALK)-expressing pulmonary adenocarcinoma. J Pathol Transl Med 2015; 49: 310-7. ArticlePubMedPMCPDF
- 25. Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res 2011; 71: 6051-60. PubMedPMC
- 26. Kim S, Kim TM, Kim DW, et al. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J Thorac Oncol 2013; 8: 415-22. ArticlePubMed
- 27. Kuo YW, Wu SG, Ho CC, Shih JY. Good response to gefitinib in lung adenocarcinoma harboring coexisting EML4-ALK fusion gene and EGFR mutation. J Thorac Oncol 2010; 5: 2039-40. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Integrative multi-omics machine learning reveals novel driver genes associations in lung adenocarcinoma
Fei Yuan, FeiMing Huang, Xiaoyu Cao, Yu-Hang Zhang, KaiYan Feng, YuSheng Bao, Tao Huang, Yu-Dong Cai
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics.2026; 1874(1): 141113. CrossRef - Ensartinib for EML4-ALK-positive lung adenocarcinoma with comorbid mutations in TP53, EGFR, and ERBB2: a case report
Xiaoqing Huang, Lingxian Zhou, Jianyong Xia, Haifeng Jian, Jinji Liu, Yunying Huang, Qingsheng Chen
Frontiers in Oncology.2025;[Epub] CrossRef - Dual EGFR L858R and KRAS G12A Mutations in Lung Adenocarcinoma: A Rare Case Report and Literature Review
Gang Wei, Jun Tang, Huaiwen Wang, Dongdong Zhang
Pharmacogenomics and Personalized Medicine.2025; Volume 18: 189. CrossRef -
Artificial Intelligence in Lung Cancer Imaging: From Data to Therapy
Michaela Cellina, Giuseppe De Padova, Nazarena Caldarelli, Dario Libri, Maurizio Cè, Carlo Martinenghi, Marco Alì, Sergio Papa, Gianpaolo Carrafiello
Critical Reviews™ in Oncogenesis.2024; 29(2): 1. CrossRef - Artificial Intelligence for Cardiothoracic Imaging: Overview of Current and Emerging Applications
Bruno Hochhegger, Romulo Pasini, Alysson Roncally Carvalho, Rosana Rodrigues, Stephan Altmayer, Leonardo Kayat Bittencourt, Edson Marchiori, Reza Forghani
Seminars in Roentgenology.2023; 58(2): 184. CrossRef - Genomic Landscape of Primary Resistance to Osimertinib Among Hispanic Patients with EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC): Results of an Observational Longitudinal Cohort Study
Diego F. Chamorro, Andrés F. Cardona, July Rodríguez, Alejandro Ruiz-Patiño, Oscar Arrieta, Darwin A. Moreno-Pérez, Leonardo Rojas, Zyanya Lucia Zatarain-Barrón, Dora V. Ardila, Lucia Viola, Gonzalo Recondo, Juan B. Blaquier, Claudio Martín, Luis Raez, Su
Targeted Oncology.2023; 18(3): 425. CrossRef - Histone deacetylase inhibitor belinostat regulates metabolic reprogramming in killing KRAS‐mutant human lung cancer cells
Rebecca M. Peter, Md. Shahid Sarwar, Sarah Z. Mostafa, Yujue Wang, Xiaoyang Su, Ah‐Ng Kong
Molecular Carcinogenesis.2023; 62(8): 1136. CrossRef - Differential Distribution of Brain Metastases from Non-Small Cell Lung Cancer Based on Mutation Status
Bihong T. Chen, Taihao Jin, Ningrong Ye, Sean W. Chen, Russell C. Rockne, Stephanie Yoon, Isa Mambetsariev, Ebenezer Daniel, Ravi Salgia
Brain Sciences.2023; 13(7): 1057. CrossRef - Clinicopathological features and prognostic significance of pulmonary adenocarcinoma with signet ring cell components: meta-analysis and SEER analysis
Yang Tan, Ying-he Huang, Jia-wen Xue, Rui Zhang, Run Liu, Yan Wang, Zhen-Bo Feng
Clinical and Experimental Medicine.2023; 23(8): 4341. CrossRef - Ganoderma microsporum immunomodulatory protein as an extracellular epidermal growth factor receptor (EGFR) degrader for suppressing EGFR-positive lung cancer cells
Wei-Jyun Hua, Hsin Yeh, Zhi-Hu Lin, Ai-Jung Tseng, Li-Chen Huang, Wei-Lun Qiu, Tsung-Hsi Tu, Ding-Han Wang, Wei-Hung Hsu, Wei-Lun Hwang, Tung-Yi Lin
Cancer Letters.2023; 578: 216458. CrossRef - Next generation sequencing for detection of EGFR alterations in NSCLC: is more better?
Ullas Batra, Shrinidhi Nathany, Mansi Sharma, Parveen Jain, Anurag Mehta
Journal of Clinical Pathology.2022; 75(3): 164. CrossRef - Enkurin domain containing 1 (ENKD1) regulates the proliferation, migration and invasion of non‐small cell lung cancer cells
Ting Song, Peng Zhou, Chunjiao Sun, Na He, Haixia Li, Jie Ran, Jun Zhou, Yue Wu, Min Liu
Asia-Pacific Journal of Clinical Oncology.2022;[Epub] CrossRef - Targeting Mutant Kirsten Rat Sarcoma Viral Oncogene Homolog in Non-Small Cell Lung Cancer: Current Difficulties, Integrative Treatments and Future Perspectives
Jia-Xin Li, Run-Ze Li, Lin-Rui Ma, Peng Wang, Dong-Han Xu, Jie Huang, Li-Qi Li, Ling Tang, Ying Xie, Elaine Lai-Han Leung, Pei-Yu Yan
Frontiers in Pharmacology.2022;[Epub] CrossRef - Histone H3K9 methyltransferase SETDB1 augments invadopodia formation to promote tumor metastasis
Shuhei Ueshima, Jia Fang
Oncogene.2022; 41(24): 3370. CrossRef - ESMO expert consensus statements on the management of EGFR mutant non-small-cell lung cancer
A. Passaro, N. Leighl, F. Blackhall, S. Popat, K. Kerr, M.J. Ahn, M.E. Arcila, O. Arrieta, D. Planchard, F. de Marinis, A.M. Dingemans, R. Dziadziuszko, C. Faivre-Finn, J. Feldman, E. Felip, G. Curigliano, R. Herbst, P.A. Jänne, T. John, T. Mitsudomi, T.
Annals of Oncology.2022; 33(5): 466. CrossRef - Molecular Targets in Lung Cancer: Study of the Evolution of Biomarkers Associated with Treatment with Tyrosine Kinase Inhibitors—Has NF1 Tumor Suppressor a Key Role in Acquired Resistance?
Begoña O. Alen, Lara S. Estévez-Pérez, María Teresa Hermida-Romero, Ana Reguera-Arias, Rosario García-Campelo, Mercedes de la Torre-Bravos, Ángel Concha
Cancers.2022; 14(14): 3323. CrossRef - Analytical and clinical validation of a custom 15-gene next-generation sequencing panel for the evaluation of circulating tumor DNA mutations in patients with advanced non-small-cell lung cancer
Yock Ping Chow, Norziha Zainul Abidin, Ken Siong Kow, Lye Mun Tho, Chieh Lee Wong, Rama Krishna Kancha
PLOS ONE.2022; 17(10): e0276161. CrossRef - Potential Therapeutic Strategy for EGFR-Mutant Lung Cancer With Concomitant EML4-ALK Rearrangement—Combination of EGFR Tyrosine Kinase Inhibitors and ALK Inhibitors
Ming-Hung Huang, Jih-Hsiang Lee, Pei-Shan Hung, James Chih-Hsin Yang
JTO Clinical and Research Reports.2022; 3(11): 100405. CrossRef - Artificial Intelligence in Lung Cancer Imaging: Unfolding the Future
Michaela Cellina, Maurizio Cè, Giovanni Irmici, Velio Ascenti, Natallia Khenkina, Marco Toto-Brocchi, Carlo Martinenghi, Sergio Papa, Gianpaolo Carrafiello
Diagnostics.2022; 12(11): 2644. CrossRef - A single center analysis of first-line treatment in advanced KRAS mutant non-small cell lung cancer: real-world practice
Yanxia Liu, Yuan Gao, Ying Wang, Cong Zhao, Zhiyun Zhang, Baolan Li, Tongmei Zhang
BMC Cancer.2022;[Epub] CrossRef - High frequency of KRAS and EGFR mutation profiles in BRAF-negative thyroid carcinomas in Indonesia
Didik Setyo Heriyanto, Vincent Laiman, Nikko Vanda Limantara, Widyan Putra Anantawikrama, Fara Silvia Yuliani, Rita Cempaka, Sumadi Lukman Anwar
BMC Research Notes.2022;[Epub] CrossRef - Kirsten Rat Sarcoma Mutation in South Indians with Non-Small Lung Cancer
Gautam Balaram, Renjan Thomas, Suhas N. Ghorpade, Prarthana V. Kowsik, Baby Dharman, Yogesh Shivakumar, Shekar Patil, Satheesh Chiradoni Thungappa, HP Shashidhara, Somorat Bhattacharjee, Sridhar Papaiah Susheela, Radheshyam Naik, Srinivas Belagutty Jayapp
Journal of Precision Oncology.2022; 2(1): 3. CrossRef - Targeted next-generation sequencing for cancer-associated gene mutation and copy number detection in 206 patients with non–small-cell lung cancer
Songbai Zheng, Xiaodan Wang, Ying Fu, Beibei Li, Jianhua Xu, Haifang Wang, Zhen Huang, Hui Xu, Yurong Qiu, Yaozhou Shi, Kui Li
Bioengineered.2021; 12(1): 791. CrossRef - How mathematical modeling could contribute to the quantification of metastatic tumor burden under therapy: insights in immunotherapeutic treatment of non-small cell lung cancer
Pirmin Schlicke, Christina Kuttler, Christian Schumann
Theoretical Biology and Medical Modelling.2021;[Epub] CrossRef - A case of concomitant EGFR/ALK alteration against a mutated EGFR background in early-stage lung adenocarcinoma
Ki-Chang Lee, Jiwon Koh, Doo Hyun Chung, Yoon Kyung Jeon
Journal of Pathology and Translational Medicine.2021; 55(2): 139. CrossRef - Malfeasance of KRAS mutations in carcinogenesis
Rupal Tripathi, Shrinidhi Nathany, Anurag Mehta, Ullas Batra, Sakshi Mattoo, Mansi Sharma
Clinical and Experimental Medicine.2021; 21(3): 439. CrossRef - Detection of Low-Frequency KRAS Mutations in cfDNA From EGFR-Mutated NSCLC Patients After First-Line EGFR Tyrosine Kinase Inhibitors
Giorgia Nardo, Jessica Carlet, Ludovica Marra, Laura Bonanno, Alice Boscolo, Alessandro Dal Maso, Andrea Boscolo Bragadin, Stefano Indraccolo, Elisabetta Zulato
Frontiers in Oncology.2021;[Epub] CrossRef - Non-Small Cell Lung Cancer Harboring Concurrent EGFR Genomic Alterations: A Systematic Review and Critical Appraisal of the Double Dilemma
Valerio Gristina, Maria La Mantia, Antonio Galvano, Sofia Cutaia, Nadia Barraco, Marta Castiglia, Alessandro Perez, Marco Bono, Federica Iacono, Martina Greco, Katia Calcara, Valentina Calò, Sergio Rizzo, Lorena Incorvaia, Maria Chiara Lisanti, Giulia San
Journal of Molecular Pathology.2021; 2(2): 173. CrossRef - Testing for EGFR Mutations and ALK Rearrangements in Advanced Non-Small-Cell Lung Cancer: Considerations for Countries in Emerging Markets
Mercedes L Dalurzo, Alejandro Avilés-Salas, Fernando Augusto Soares, Yingyong Hou, Yuan Li, Anna Stroganova, Büge Öz, Arif Abdillah, Hui Wan, Yoon-La Choi
OncoTargets and Therapy.2021; Volume 14: 4671. CrossRef - Untangling the KRAS mutated lung cancer subsets and its therapeutic implications
Kulshrestha Ritu, Pawan Kumar, Amit Singh, K. Nupur, Sonam Spalgias, Parul Mrigpuri, Rajkumar
Molecular Biomedicine.2021;[Epub] CrossRef - Lorlatinib Induces Durable Disease Stabilization in a Pancreatic Cancer Patient with a ROS1 p.L1950F Mutation: Case Report
Janna-Lisa Velthaus, Peter Iglauer, Ronald Simon, Carsten Bokemeyer, Peter Bannas, Niklas Beumer, Charles D. Imbusch, Eray Goekkurt, Sonja Loges
Oncology Research and Treatment.2021; 44(9): 495. CrossRef - Proteasome-dependent degradation of Smad7 is critical for lung cancer metastasis
Lu Tong, Shihui Shen, Quan Huang, Junjiang Fu, Tianzhen Wang, Linian Pan, Pei Zhang, Geng Chen, Tingmei Huang, Ke Li, Qingwu Liu, Shaofang Xie, Xiao Yang, Robb E. Moses, Xiaotao Li, Lei Li
Cell Death & Differentiation.2020; 27(6): 1795. CrossRef - Circulating Cell-Free Dna As A Diagnostic and Prognostic Biomarker for Non-Small-Cell Lung Cancer: A Systematic Review and Meta-Analysis
Zhoumiao Chen, Huiwen Miao, Qingxin Zeng, Shaohua Xu, Zhao Chen, Kai Liu
Biomarkers in Medicine.2020; 14(7): 587. CrossRef - Influence of EGFR-activating mutations on sensitivity to tyrosine kinase inhibitors in a KRAS mutant non-small cell lung cancer cell line
Yoshinori Tsukumo, Mikihiko Naito, Takayoshi Suzuki, Srikumar Chellappan
PLOS ONE.2020; 15(3): e0229712. CrossRef - KRAS oncogene may be another target conquered in non‐small cell lung cancer (NSCLC)
Hanxiao Chen, Jun Zhao
Thoracic Cancer.2020; 11(12): 3425. CrossRef - Epidemiologic Features of NSCLC Gene Alterations in Hispanic Patients from Puerto Rico
Ruifang Zheng, Zhiwei Yin, Albert Alhatem, Derek Lyle, Bei You, Andrew S. Jiang, Dongfang Liu, Zsolt Jobbagy, Qing Wang, Seena Aisner, Jie-Gen Jiang
Cancers.2020; 12(12): 3492. CrossRef - Comprehensive pancancer genomic analysis reveals (RTK)-RAS-RAF-MEK as a key dysregulated pathway in cancer: Its clinical implications
Robin Imperial, Omer M Toor, Arif Hussain, Janakiraman Subramanian, Ashiq Masood
Seminars in Cancer Biology.2019; 54: 14. CrossRef - Epidermal growth factor receptor (EGFR), KRAS, and BRAF mutations in lung adenocarcinomas: A study from India
Varsha Singh, Prerna Guleria, Prabhat Singh Malik, Anant Mohan, Sanjay Thulkar, R M Pandey, Kalpana Luthra, Sudheer Arava, Ruma Ray, Deepali Jain
Current Problems in Cancer.2019; 43(5): 391. CrossRef - Clinical Validation of Coexisting Activating Mutations Within EGFR, Mitogen-Activated Protein Kinase, and Phosphatidylinositol 3-Kinase Pathways in Lung Cancers
Federico De Marchi, Lisa Haley, Henderson Fryer, Junaid Ibrahim, Katie Beierl, Gang Zheng, Christopher D. Gocke, James R. Eshleman, Deborah Belchis, Peter Illei, Ming-Tseh Lin
Archives of Pathology & Laboratory Medicine.2019; 143(2): 174. CrossRef - A sequential Monte Carlo algorithm for inference of subclonal structure in cancer
Oyetunji E. Ogundijo, Kaiyi Zhu, Xiaodong Wang, Dimitris Anastassiou, Xiang Li
PLOS ONE.2019; 14(1): e0211213. CrossRef - The Presence of Concomitant Mutations Affects the Activity of EGFR Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer (NSCLC) Patients
Anna Rachiglio, Francesca Fenizia, Maria Piccirillo, Domenico Galetta, Lucio Crinò, Bruno Vincenzi, Emiddio Barletta, Carmine Pinto, Francesco Ferraù, Matilde Lambiase, Agnese Montanino, Cristin Roma, Vienna Ludovini, Elisabetta Montagna, Antonella De Luc
Cancers.2019; 11(3): 341. CrossRef - Concurrent Driver Gene Mutations as Negative Predictive Factors in Epidermal Growth Factor Receptor-Positive Non-Small Cell Lung Cancer
Minjiang Chen, Yan Xu, Jing Zhao, Wei Zhong, Li Zhang, Yalan Bi, Mengzhao Wang
EBioMedicine.2019; 42: 304. CrossRef - Anaplastic Lymphoma Kinase (ALK)-positive Tumors
Rohan Gupta, Idoroenyi Amanam, Syed Rahmanuddin, Isa Mambetsariev, Yingyu Wang, Charity Huang, Karen Reckamp, Lalit Vora, Ravi Salgia
American Journal of Clinical Oncology.2019; 42(4): 337. CrossRef - Clinical features and therapeutic options in non‐small cell lung cancer patients with concomitant mutations of EGFR, ALK, ROS1, KRAS or BRAF
Xibin Zhuang, Chao Zhao, Jiayu Li, Chunxia Su, Xiaoxia Chen, Shengxiang Ren, Xuefei Li, Caicun Zhou
Cancer Medicine.2019; 8(6): 2858. CrossRef - Intrinsic Resistance to EGFR-Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: Differences and Similarities with Acquired Resistance
Eric Santoni-Rugiu, Linea C. Melchior, Edyta M. Urbanska, Jan N. Jakobsen, Karin de Stricker, Morten Grauslund, Jens B. Sørensen
Cancers.2019; 11(7): 923. CrossRef - Clinical outcome of treatment of metastatic non-small cell lung cancer in patients harboring uncommon EGFR mutation
J. Chantharasamee, N. Poungvarin, P. Danchaivijitr, S. Techawatanawanna
BMC Cancer.2019;[Epub] CrossRef - Deregulated lncRNA expression profile in the mouse lung adenocarcinomas with KRAS‐G12D mutation and P53 knockout
Meiqin Zhang, Nan Jiang, Renjie Cui, Sichen Du, Huayuan Ou, Tinglan Chen, Runsheng Ge, Duan Ma, Jin Zhang
Journal of Cellular and Molecular Medicine.2019; 23(10): 6978. CrossRef - Open-Sourced CIViC Annotation Pipeline to Identify and Annotate Clinically Relevant Variants Using Single-Molecule Molecular Inversion Probes
Erica K. Barnell, Adam Waalkes, Matt C. Mosior, Kelsi Penewit, Kelsy C. Cotto, Arpad M. Danos, Lana M. Sheta, Katie M. Campbell, Kilannin Krysiak, Damian Rieke, Nicholas C. Spies, Zachary L. Skidmore, Colin C. Pritchard, Todd A. Fehniger, Ravindra Uppalur
JCO Clinical Cancer Informatics.2019; (3): 1. CrossRef - EGFR, KRAS, BRAF, ALK, and cMET genetic alterations in 1440 Sardinian patients with lung adenocarcinoma
Maria Colombino, Panagiotis Paliogiannis, Antonio Cossu, Davide Adriano Santeufemia, Maria Cristina Sini, Milena Casula, Grazia Palomba, Antonella Manca, Marina Pisano, Valentina Doneddu, Giuseppe Palmieri
BMC Pulmonary Medicine.2019;[Epub] CrossRef - Concomitant Presence of EGFR and ALK Fusion Gene Mutation in Adenocarcinoma of Lung: A Case Report and Review of the Literature
Nishitha Thumallapally, Hana Yu, Mohammad Farhan, Uroosa Ibrahim, Maricel Odiami
Journal of Pharmacy Practice.2018; 31(2): 244. CrossRef - Double Trouble: A Case Series on Concomitant Genetic Aberrations in NSCLC
Nele Van Der Steen, Yves Mentens, Marc Ramael, Leticia G. Leon, Paul Germonpré, Jose Ferri, David R. Gandara, Elisa Giovannetti, Godefridus J. Peters, Patrick Pauwels, Christian Rolfo
Clinical Lung Cancer.2018; 19(1): 35. CrossRef - KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target
Marta Román, Iosune Baraibar, Inés López, Ernest Nadal, Christian Rolfo, Silvestre Vicent, Ignacio Gil-Bazo
Molecular Cancer.2018;[Epub] CrossRef - Pulmonary Adenocarcinoma, Harboring Both an EGFR Mutation and ALK Rearrangement, Presenting a Stable Disease to Erlotinib and a Partial Response to Alectinib
Akira Yokoyama, Atsuhisa Tamura, Kazuko Miyakawa, Kei Kusaka, Masahiro Shimada, Takashi Hirose, Hirotoshi Matsui, Masashi Kitani, Akira Hebisawa, Ken Ohta
Internal Medicine.2018; 57(16): 2377. CrossRef - Long-term survival with erlotinib in advanced lung adenocarcinoma harboring synchronous EGFR G719S and KRAS G12C mutations
Biagio Ricciuti, Sara Baglivo, Vienna Ludovini, Angelo Sidoni, Giulio Metro, Marta Brambilla, Annamaria Siggillino, Maria Sole Reda, Alberto Rebonato, Daniele Maiettini, Rita Chiari
Lung Cancer.2018; 120: 70. CrossRef - Concomitant driver mutations in advanced EGFR-mutated non-small-cell lung cancer and their impact on erlotinib treatment
Jan Nyrop Jakobsen, Eric Santoni-Rugiu, Morten Grauslund, Linea Melchior, Jens Benn Sørensen
Oncotarget.2018; 9(40): 26195. CrossRef - Implications of KRAS mutations in acquired resistance to treatment in NSCLC
Marzia Del Re, Eleonora Rofi, Giuliana Restante, Stefania Crucitta, Elena Arrigoni, Stefano Fogli, Massimo Di Maio, Iacopo Petrini, Romano Danesi
Oncotarget.2018; 9(5): 6630. CrossRef - Alk Immunohistochemistry is Highly Sensitive and Specific for the Detection of Alk Translocated Lung Adenocarcinomas: Lessons from An Audit of Lung Cancer Molecular Testing
YC Kheng, K Walsh, L Williams, WA Wallace, DJ Harrison, A Oniscu
Journal of the Royal College of Physicians of Edinburgh.2018; 48(1): 20. CrossRef - Targeting KRAS mutated non-small cell lung cancer: A history of failures and a future of hope for a diverse entity
Alexios Matikas, Dimitrios Mistriotis, Vasilios Georgoulias, Athanasios Kotsakis
Critical Reviews in Oncology/Hematology.2017; 110: 1. CrossRef - Clinical Outcome of ALK -Positive Non–Small Cell Lung Cancer (NSCLC) Patients with De Novo EGFR or KRAS Co-Mutations Receiving Tyrosine Kinase Inhibitors (TKIs)
Sabine Schmid, Oliver Gautschi, Sacha Rothschild, Michael Mark, Patrizia Froesch, Dirk Klingbiel, Hermann Reichegger, Wolfram Jochum, Joachim Diebold, Martin Früh
Journal of Thoracic Oncology.2017; 12(4): 681. CrossRef - EGFR and KRAS molecular genotyping for pulmonary carcinomas: Feasibility of a simple and rapid technique implementable in any department of pathology
Vincent Thomas De Montpréville, Maria-Rosa Ghigna, Ludovic Lacroix, Antoinette Lemoine, Benjamin Besse, Olaf Mercier, Élie Fadel, Peter Dorfmuller, Thierry Le Chevalier
Pathology - Research and Practice.2017; 213(7): 793. CrossRef - MET Exon 14 Skipping Mutations in Lung Adenocarcinoma: Clinicopathologic Implications and Prognostic Values
Geun Dong Lee, Seung Eun Lee, Doo-Yi Oh, Dan-bi Yu, Hae Min Jeong, Jooseok Kim, Sungyoul Hong, Hun Soon Jung, Ensel Oh, Ji-Young Song, Mi-Sook Lee, Mingi Kim, Kyungsoo Jung, Jhingook Kim, Young Kee Shin, Yoon-La Choi, Hyeong Ryul Kim
Journal of Thoracic Oncology.2017; 12(8): 1233. CrossRef - Non-Small-Cell Lung Cancer (NSCLC) Harboring ALK Translocations: Clinical Characteristics and Management in a Real-Life Setting: a French Retrospective Analysis (GFPC 02–14 Study)
Jean-Bernard Auliac, Isabelle Monnet, Catherine Dubos-Arvis, Anne Marie Chiappa, Nathalie Baize, Suzana Bota, Alain Vergnenegre, Helene Doubre, Chrystele Locher, Acya Bizieux, Gilles Robinet, Christos Chouaid
Targeted Oncology.2017; 12(6): 833. CrossRef - Concomitant EML4-ALK rearrangement and EGFR mutation in non-small cell lung cancer patients: a literature review of 100 cases
Giuseppe Lo Russo, Martina Imbimbo, Giulia Corrao, Claudia Proto, Diego Signorelli, Milena Vitali, Monica Ganzinelli, Laura Botta, Nicoletta Zilembo, Filippo de Braud, Marina Chiara Garassino
Oncotarget.2017; 8(35): 59889. CrossRef - Management of non-small cell lung cancer in the era of personalized medicine
Gaetano Rocco, Alessandro Morabito, Alessandra Leone, Paolo Muto, Francesco Fiore, Alfredo Budillon
The International Journal of Biochemistry & Cell Biology.2016; 78: 173. CrossRef - A long-term survivor of non-small-cell lung cancer harboring concomitant EGFR mutation and ALK translocation
Fumio Imamura, Takako Inoue, Madoka Kimura, Kazumi Nishino, Toru Kumagai
Respiratory Medicine Case Reports.2016; 19: 137. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
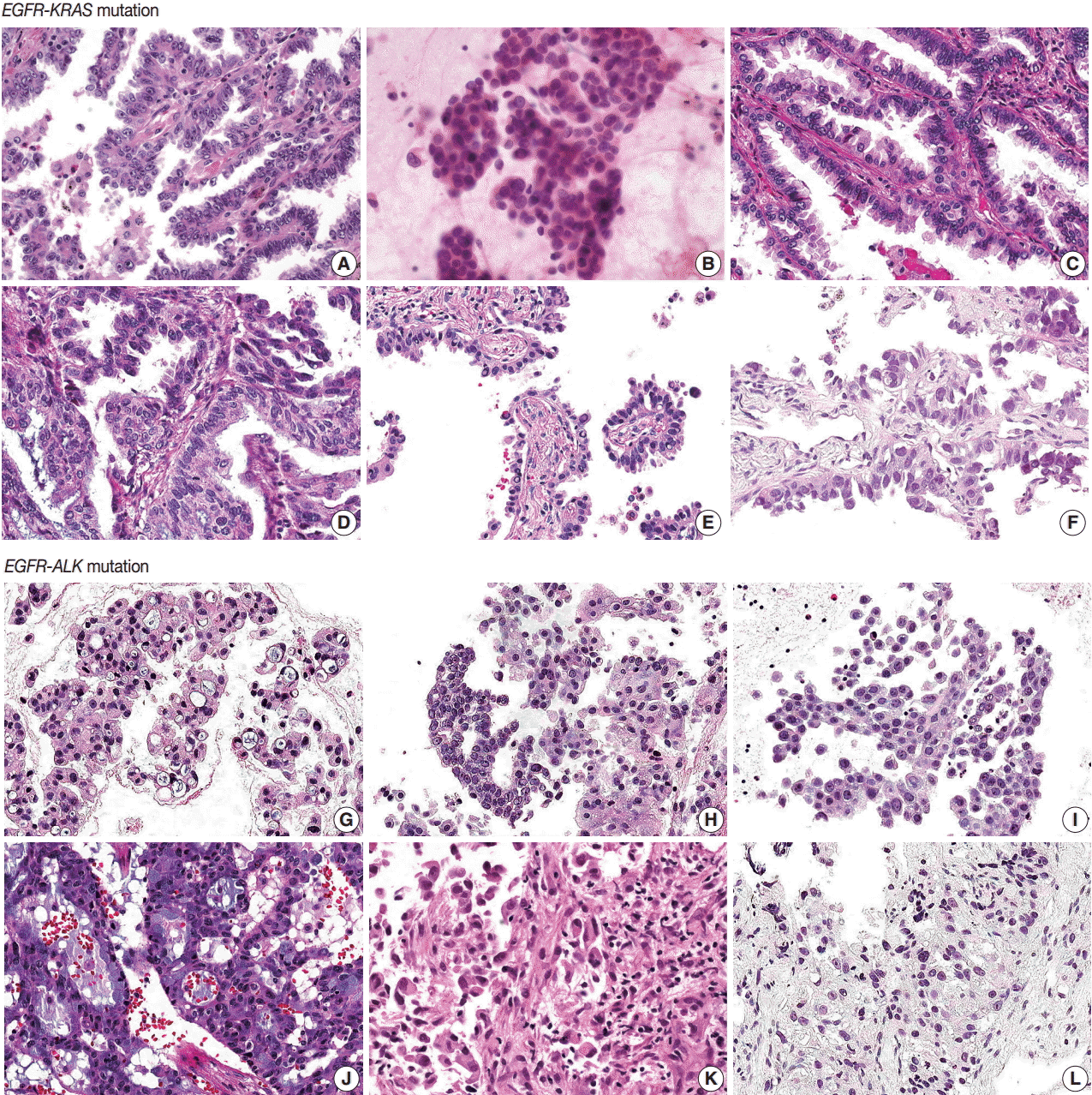

Fig. 1.
| Patient No. | Sex/Age(yr) | Smoking | Specimen | Stage | EGFR mutation | Additional mutation | Dominant pattern | Typical cell feature | Mucin | Intranuclear inclusion | Surgery | Targeted therapy | Response | Status | PFS (mo) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M/74 | 57PY | Lung/R | pT1bN1 | E21 L858R | KRAS I21S | Papillary and micropapillary | Hobnail cells | No | No | + | - | - | DOD |
- |
| 2 | F/62 | Never | Lung/A | M1(IV) | E21 L858R | KRAS G12D | No | No | Present | Gefitinib | PR → PD | DOD | 7 | ||
| 3 | F/63 | Never | Lung/R | pT1bNO | E21 L858R | KRAS G13A | Papillary and acinar | Hobnail cells | No | No | + | - | - | NED | - |
| 4 | F/48 | Never | Lung/R | pT1bN2 | E19 deletion | KRAS G12V | Acinar and solid | Hobnail and columnar cells | Intra- and extracytoplasmic | No | + | Erlotinib | PR → PD | DOD | 20 |
| 5 | M/55 | 20PY | Lung/R | pT2N2 | E19 deletion | KRAS G13C | Solid and acinar | Hobnail cells | No | Present | + | Gefitinib | PR | AWD | 29 |
| 6 | F/78 | Never | Lung/B | M1(IV) | E18 G719X | KRAS G12D | Papillary and acinar | Hobnail cells | No | No | - | (refused) | - | AWD |
- |
| 7 | F/62 | Never | LN/B | M1(IV) | E21 L858R |
ALK | Solid and cribriform | Signet ring cells | Intracytoplasmic | No | + |
Gefitinib | PR | AWD |
18 |
| 8 | F/62 | Never | Lung/B | M1(IV) | E21 L858R | ALK | Solid and cribriform | No | No | No | - | Gefitinib and Crizotinib | SD and PR | AWD | 24 |
| E18 G719X | |||||||||||||||
| 9 | M/56 | 15PY | Lung/B | M1(IV) | E18 G719X |
ALK | Solid | No | No | No | Crizotinib | SD | DOD | 4 | |
| 10 | F/68 | Never | Lung/R | ypT2N2 | E18 G719X |
ALK | Solid, micropapillary and cribriform | Signet ring cells | Intra- and extracytoplasmic | Present | + | - | - | NED | - |
| 11 | F/58 | Never | Lung/B | M1(IV) | E19 deletion | ALK | Solid | Signet ring cells | Intracytoplasmic | No | - | Crizotinib | - | AWD |
|
| 12 | F/66 | Never | Adrenal/B | M1(IV) | E20 R803W | ALK | Solid | No | No | No | + |
Erlotinib | PD | AWD |
0.7 |
EGFR, epidermal growth factor receptor; PFS, progression-free survival; M, male; PY, pack-year; R, resection; E, exon; KRAS, v-Ki-ras2 Kirsten rat sarcoma viral oncogene; DOD, died of disease; F, female; A, aspiration; PR, partial response; PD, progressive disease; NED, no evidence of disease; AWD, alive with disease; B, biopsy; LN, lymph node; ALK, anaplastic lymphoma kinase; SD, stable disease. Lost to follow-up; Cannot identify dominant architectural patterns in an aspiration slide; Mutation detected only in PNA clamping method; Have a history of lobectomy at outside hospital, prior to biopsy of metastatic lesion; Cannot evaluate due to lost to follow-up.

