 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 52(1); 2018 > Article
-
Original Article
The Smad4/PTEN Expression Pattern Predicts Clinical Outcomes in Colorectal Adenocarcinoma - Yumin Chung, Young Chan Wi, Yeseul Kim, Seong Sik Bang, Jung-Ho Yang1, Kiseok Jang, Kyueng-Whan Min, Seung Sam Paik
-
Journal of Pathology and Translational Medicine 2018;52(1):37-44.
DOI: https://doi.org/10.4132/jptm.2017.10.20
Published online: October 23, 2017
Department of Pathology, Hanyang University College of Medicine, Seoul, Korea
1Department of Pathology, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea
-
Corresponding Author Kyueng-Whan Min, MD, PhD Department of Pathology, Hanyang University Guri Hospital, Hanyang University College of Medicine, 153 Gyeongchun-ro, Guri 11923, Korea Tel: +82-31-560-2496 Fax: +82-31-560-2339 E-mail: kyueng@gmail.com
Seung Sam Paik, MD, PhD Department of Pathology, Hanyang University Seoul Hospital, Hanyang University College of Medicine, 222-1 Wangsimni-ro, Seongdong-gu, Seoul 04763, Korea Tel: +82-2-2290-8252 Fax: +82-2-2296-7502 E-mail: sspaik@hanyang.ac.kr
© 2018 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background
- Smad4 and PTEN are prognostic indicators for various tumor types. Smad4 regulates tumor suppression, whereas PTEN inhibits cell proliferation. We analyzed and compared the performance of Smad4 and PTEN for predicting the prognosis of patients with colorectal adenocarcinoma.
-
Methods
- Combined expression patterns based on Smad4+/– and PTEN+/– status were evaluated by immunostaining using a tissue microarray of colorectal adenocarcinoma. The relationships between the protein expression and clinicopathological variables were analyzed.
-
Results
- Smad4–/PTEN– status was most frequently observed in metastatic adenocarcinoma, followed by primary adenocarcinoma and tubular adenoma (p<.001). When Smad4–/PTEN– and Smad4+/PTEN+ groups were compared, Smad4–/PTEN– status was associated with high N stage (p=.018) and defective mismatch repair proteins (p=.006). Significant differences in diseasefree survival and overall survival were observed among the three groups (Smad4+/PTEN+, Smad4–/PTEN+ or Smad4+/PTEN–, and Smad4–/PTEN–) (all p<.05).
-
Conclusions
- Concurrent loss of Smad4 and PTEN may lead to more aggressive disease and poor prognosis in patients with colorectal adenocarcinoma compared to the loss of Smad4 or PTEN alone.
- Patients and tissue specimens
- This retrospective study enrolled a consecutive series of 529 patients with colorectal adenocarcinoma, 48 patients with metastatic adenocarcinoma, 14 patients with tubular adenoma, and 16 patients with normal colonic mucosae. All patients were diagnosed and treated at our institute from January 1991 to August 2001. This study included data obtained from a previously conducted research study [15]. The Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK) criteria were followed throughout this study. Inclusion criteria included histopathological evidence of adenocarcinoma confirmed by a pathologist and known clinical outcome. Exclusion criteria included diagnosis of adenocarcinoma with neoadjuvant chemotherapy or inadequate clinical history. Clinicopathological findings were collected from the patients’ medical records and pathological reports. Patient age ranged from 17 to 87 years (mean, 57.7 years). The tumors were located at the cecum (n = 18), ascending colon (n = 78), hepatic flexure (n = 12), transverse colon (n=26), splenic flexure (n = 4), descending colon (n = 25), sigmoid colon (n = 113), and rectum (n = 253). Tumor size ranged from 3 to 150 mm (mean, 57 mm). A mean number of 26.7 lymph nodes (LN) were dissected for each specimen. Over a mean follow-up interval after surgery of 5.9 years, 183 patients (34.6%) died and 346 (65.4%) survived. Normal colonic mucosae, tubular adenomas, and metastatic adenocarcinoma tissues were randomly selected for evaluation of Smad4 and PTEN expression.
- This study (involving human participants) was approved by the Ethics Committee of Hanyang University Hospital (No. 2016-12-030-001) and was performed in accordance with the ethical standards of the Declaration of Helsinki, as revised in 2008. The institutional review board review confirmed that informed consent was not necessary for this study.
- Tissue microarray construction
- A representative area was carefully selected and marked on a hematoxylin and eosin (H&E)-stained slide. Tissue microarrays (TMAs) were assembled using a tissue-array instrument (Quick-Ray Manual Tissue Microarrayer, Unitma Co. Ltd., Seoul, Korea) consisting of thin-walled stainless steel punches and stylets used to empty and transfer the needle content. Areas rich in tumor cells were identified by light microscopic examination of H&Estained sections and selected for use in TMAs. Tissue cylinders with a diameter of 2 mm were punched from the previously marked tumor area (tumor center) of each donor block and transferred to a recipient paraffin block. All tissue cores were composed of tumor tissue in more than 70% of the core area. Considering the limitations associated with obtaining representative areas of a tumor, we used a 2-mm tissue core from each donor block.
- Immunohistochemical staining and scoring of Smad4 and PTEN expression
- Primary mouse monoclonal anti-Smad4 antibodies (clone B-8, Santa Cruz Biotechnology Inc, Heidelberg, Germany) were diluted 1:250 and primary mouse monoclonal anti-PTEN antibodies (clone 28H6, Novocastra Laboratories, Newcastle upon Tyne, UK) were diluted 1:200.
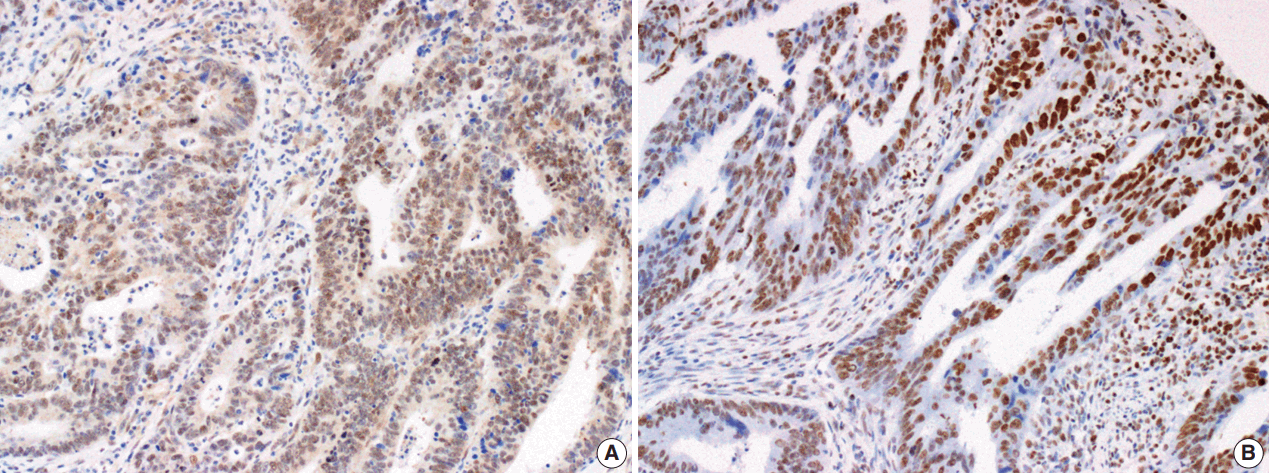
- The Smad4 and PTEN immunohistochemical (IHC) staining results were interpreted by two pathologists (K.J. and S.S.P.) who were blinded to the clinical data. Discordant cases were reviewed on a multi-headed microscope to reach consensus. Each slide was interpreted semi-quantitatively according to the intensity of nuclear immunoreactivity (Fig. 1). The cut-off was determined by proportion rather than by intensity, because the latter was rather heterogeneous. Smad4 and PTEN expression was classified as negative (< 10% positive tumor cells out of all cancer cells) or positive (≥ 10% positive tumor cells out of all cancer cells) [29].
- Statistical analysis
- The chi-square test was used to evaluate potential associations between Smad4 expression/PTEN expression and clinicopathologic parameters including age, gender, tumor location, tumor size, growth pattern, histological grade, of American Joint Committee on Cancer (AJCC) stage (T and N criteria), and MMR protein expression. The relationships between Smad4 and PTEN expression and normal mucosae, adenomatous polyps, adenocarcinomas, and metastatic LNs were analyzed using linear-by-linear association. Overall survival (OS) was defined as the time from curative surgery to death from any cause. Diseasefree survival (DFS) was defined as the time from curative surgery to the first tumor recurrence or distant metastasis. Kaplan–Meier curves with the log-rank test were used to calculate OS and DFS. Multivariate survival analysis was used with a Cox proportional hazards regression model to evaluate independent prognostic factors. A p-value of < .05 was considered significant. Statistical analysis was performed using SPSS ver. 20.0 (IBM Corp., Armonk, NY, USA) or R packages (http://www.r-project.org/).
MATERIALS AND METHODS
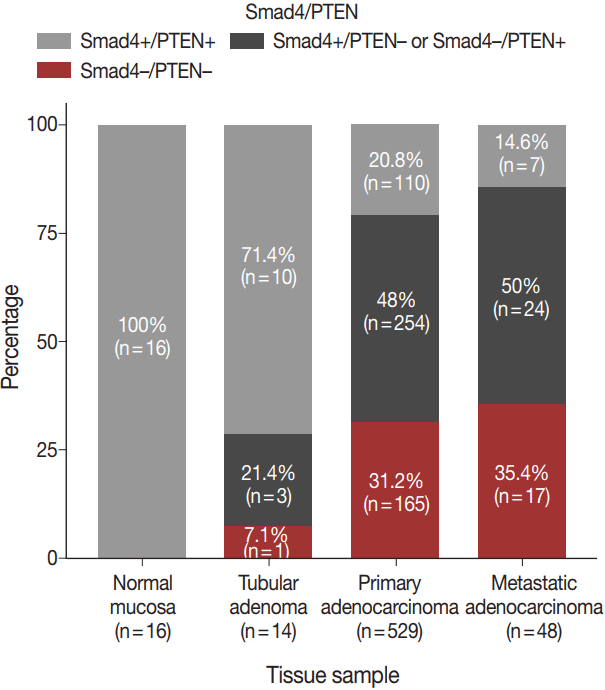
- The expression patterns of Smad4 and PTEN were evaluated in normal colonic mucosae, tubular adenomas, primary adenocarcinomas, and metastatic adenocarcinomas. The loss of both Smad4 and PTEN was associated with cancer progression. Specifically, Smad4–/PTEN– status was not observed in any of the 16 normal colonic mucosa tissues; however, it was observed in one of 14 tubular adenomas (7.1%), 165 of 529 primary adenocarcinomas (31.2%), and 17 of 48 metastatic adenocarcinomas (35.4%) (p < .001) (Fig. 2).
- Smad4 loss was associated with higher N stage (p = .013) and showed a tendency to correlate with lymphovascular invasion (p = .081) (Supplementary Table S1). PTEN loss was correlated with defective MMR (p = .005) (Supplementary Table S2).
- The distribution of Smad4 and PTEN expression status was as follows: Smad4–/PTEN–, 165 patients; Smad4–/PTEN+, 155 patients; Smad4+/PTEN–, 99 patients; and Smad4+/PTEN+, 110 patients.
- Comparison of the Smad4–/PTEN– and Smad4+/PTEN+ groups showed that Smad4–/PTEN– status was associated with higher N stage (p = .018) and defective MMR (p = .006) (Table 1). In a comparative analysis between the Smad4+/PTEN+ and Smad4–/PTEN+ or Smad4+/PTEN– groups, Smad4+/PTEN+ status was correlated with lower N stage (p = .047), absence of lymphovascular invasion (p = .049), and intact MMR (p = .006) (Supplementary Table S3). Another comparative analysis between the Smad4–/PTEN+ or Smad4+/PTEN– and Smad4–/PTEN– groups showed that Smad4–/PTEN– status was correlated with defective MMR (p = .030) (Supplementary Table S4).
- Survival according to Smad4/PTEN expression
- Univariate and multivariate survival analyses showed that the loss of Smad4 was associated with short DFS and OS (all p < .05). In univariate analyses, loss of PTEN was not associated with short DFS or OS (all p < .05). In multivariate analyses, no relationship was observed between DFS or OS and PTEN.
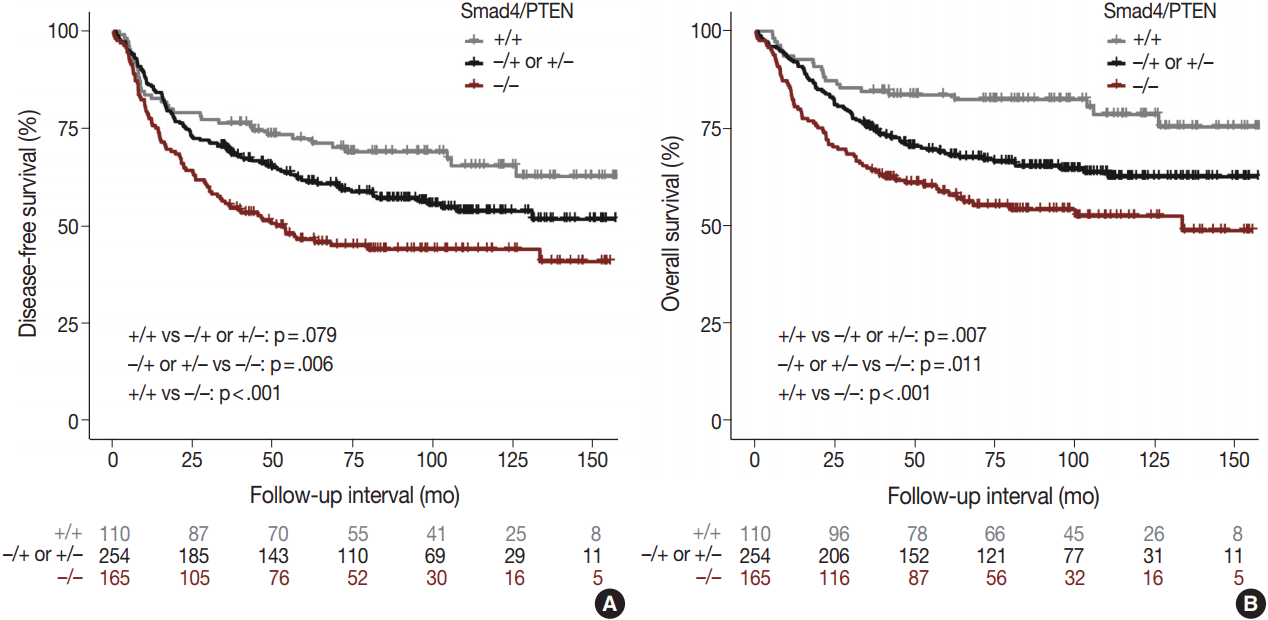
- Comparison of the Smad4+/PTEN+ and Smad4–/PTEN– groups showed that Smad4–/PTEN– status was associated with short DFS and OS in both univariate and multivariate analyses (all p < .05). Comparison of the Smad4–/PTEN+ or Smad4+/PTEN– and Smad4–/PTEN– groups revealed significant differences in DFS and OS (all p <. 05). Comparison of the Smad4+/PTEN+ and Smad4–/PTEN+ or Smad4+/PTEN– groups showed a significant difference in OS (p = .035), whereas the difference in DFS was not significant (Table 2, Fig. 3).
RESULTS
- The aim of this study was to determine whether the Smad4/PTEN expression pattern is associated with clinical outcomes. Smad4 is part of a protein complex linked to signal transduction and interacts with DNA for cell proliferation [30]. PTEN is a negative regulator of the Akt signaling pathway and is necessary for cell proliferation and inhibition of apoptosis [31,32]. Smad4 and PTEN have been shown to be closely linked in the regulation of tumor invasion and distant metastasis. For instance, Haeger et al. [33] demonstrated that the loss of Smad4 could initiate lung cancer development from keratinized epithelium in vivo. Moreover, loss of Smad4 expression and low Smad4 expression were both shown to increase cell proliferation secondary to the loss of TGF-β– mediated growth suppression [34]. During cancer development, deletion of PTEN is common in several malignancies such as gastric, esophageal, endometrial, breast, and non-small cell lung cancers [25,35-40]. Moreover, many studies have identified PTEN mutations in HRS320 cells, a colonic cancer cell line, and shown that these mutations are significantly related to poor clinical outcomes in patients with CRC [41,42].
- The prognostic value of combinations of cancer markers for patients with CRC (which exhibits high heterogeneity) may be better than that of models using a single marker. An in vivo study demonstrated that loss of both Smad4 and PTEN may act synergistically to regulate cell proliferation and accelerate tumorigenesis [25]. The synergistic action of these genes is mediated by concomitant repression of PTEN transcription by Smad4 and promotion of Smad4 degradation by PTEN [22]. Liu et al. [27] showed that the combined loss of Smad4 and PTEN was linked to metastatic lung cancer through activation of the ErbB2/ELF3/AKT pathway. In pancreatic cancer, the absence of Smad4 and PTEN has been shown to synergistically promote ductal carcinoma [28]. Intriguingly, Smad4 deletion has been shown to drive progression of PTEN-deficient prostate cancer to highly aggressive behaviors such as tumor invasion and lymph node metastasis [24]. Thus, Smad4 deficiency can accelerate tumor cell proliferation and invasion, especially when combined with loss of PTEN expression. Although this relationship has been confirmed in an in vitro study, no reports have described a difference in DFS or OS in human CRC according to the loss of both Smad4 and PTEN. In the present study, we showed that Smad4+/PTEN+ status indicated good prognosis, whereas Smad4−/PTEN– status was associated with poor prognosis. These findings suggest that the loss of both Smad4 and PTEN expression is linked to worse clinical outcomes in patients with CRC. In addition, Smad4–/PTEN– status was significantly associated with higher N stage compared to Smad4+/PTEN+ status.
- The precise mechanism underlying the synergistic effect between Smad4 and PTEN is not fully understood. In esophageal cancer, it has been proposed that the loss of both Smad4 and PTEN leads to the downregulation of CIP/KIP and INK4 Cdk inhibitors and subsequent suppression of cell cycle arrest [43]. Alternatively, concurrent deletion of PTEN and Smad4 has been reported to induce an oncogenic pathway including ErbB2/Akt/ELF3 and to repress tumor suppressors in lung cancer [27]. Interestingly, the lung cancer study demonstrated that the mRNA level of ELF3 (a transcriptional factor known to have oncogenic activity) was significantly higher in Smad4–/PTEN– mice compared to Smad4– mice and PTEN– mice. In pancreatic cancer, Smad4–/PTEN– status was associated with wider cancer spread and larger tumors compared with Smad4– status or PTEN– status [28]. In the present study, the Smad4–/PTEN+ and Smad4+/PTEN– groups had lower survival rates than the Smad4+/PTEN+ group, but higher survival rates than the Smad4–/PTEN– group. These biological processes, together with the concurrent loss of Smad4 and PTEN, may correlate with more aggressive clinical behavior than that seen with the loss of a single protein.
- This study did have some limitations. First, in contrast to previous in vitro studies, we did not demonstrate a molecular interaction between Smad4 and PTEN, making it difficult to draw concrete conclusions from our data. Second, the cut-off value of 10% for distinguishing positive from negative expression of Smad4 and PTEN was determined by IHC evaluation. However, Smad4 and PTEN distribution may vary according to specimen type, implying that this cut-off point may not apply to other types of clinical samples or to other laboratories. Third, interactions between Smad4 and PTEN within single tumor cells could not be observed, because double staining of Smad4 and PTEN expression was not performed.
- In summary, we found that the Smad4/PTEN expression pattern was statistically correlated with several clinicopathological parameters and with survival. Smad4–/PTEN– status was more frequently observed in patients who followed the multi-step carcinogenesis progression from normal colonic mucosae to tubular adenoma, primary adenocarcinoma, and metastatic adenocarcinoma. Some differences in survival were observed among the Smad4+/PTEN+, Smad4+/PTEN– or Smad4–/PTEN+, and Smad4–/PTEN– groups. Thus, combinations of markers may be more useful for predicting the survival of patients with CRC than single markers alone. We conclude that concurrent loss of Smad4 and PTEN may accelerate cancer progression and that these two genes thus represent potential targets for cancer treatment.
DISCUSSION
Electronic Supplementary Material
| Parameter | No. (n = 275) | Smad4+/PTEN+ (n = 110) | Smad4–/PTEN– (n = 165) | p-value (chi-square test) |
|---|---|---|---|---|
| Age (yr) | .367 | |||
| < 65 | 189 | 79 (71.8) | 110 (66.7) | |
| ≥ 65 | 86 | 31 (28.2) | 55 (33.3) | |
| Sex | .519 | |||
| Female | 121 | 51 (46.4) | 70 (42.4) | |
| Male | 154 | 59 (53.6) | 95 (57.6) | |
| Tumor location | .413a | |||
| Right | 63 | 27 (24.5) | 36 (21.8) | |
| Transverse and left | 25 | 12 (10.9) | 13 (7.9) | |
| Rectosigmoid | 187 | 71 (64.5) | 116 (70.3) | |
| Tumor size (cm) | .472 | |||
| ≤ 4.5 | 98 | 42 (38.2) | 56 (33.9) | |
| > 4.5 | 177 | 68 (61.8) | 109 (66.1) | |
| Growth pattern | .866 | |||
| Fungating | 71 | 29 (26.4) | 42 (25.5) | |
| Ulcerating | 204 | 81 (73.6) | 123 (74.5) | |
| Histologic grade | .726a | |||
| 1 | 13 | 3 (2.7) | 10 (6.1) | |
| 2 | 200 | 83 (75.5) | 117 (70.9) | |
| 3 | 62 | 24 (21.8) | 38 (23) | |
| AJCC stage | .135a | |||
| Tis | 5 | 1 (0.9) | 4 (2.4) | |
| I | 20 | 12 (10.9) | 8 (4.8) | |
| II | 101 | 44 (40) | 57 (34.5) | |
| III | 149 | 53 (48.2) | 96 (58.2) | |
| T stage | .251a | |||
| Tis | 5 | 1 (0.9) | 4 (2.4) | |
| 1 | 007 | 5 (4.5) | 2 (1.2) | |
| 2 | 22 | 12 (10.9) | 10 (6.1) | |
| 3 | 231 | 89 (80.9) | 142 (86.1) | |
| 4 | 10 | 3 (2.7) | 7 (4.2) | |
| N stage | .018*a | |||
| 0 | 126 | 57 (51.8) | 69 (41.8) | |
| 1 | 67 | 30 (27.3) | 37 (22.4) | |
| 2 | 82 | 26 (35.8) | 59 (35.8) | |
| Lymphovascular invasion | .138 | |||
| Absence | 125 | 56 (50.9) | 69 (41.8) | |
| Presence | 150 | 54 (49.1) | 96 (58.2) | |
| Mismatch repair protein | .006* | |||
| Intact | 147 | 70 (63.6) | 77 (46.7) | |
| Defective | 128 | 40 (36.4) | 88 (53.3) |
| Univariate significance | Multivariate significance | HR | 95% CI | |
|---|---|---|---|---|
| p-valuea | p-valueb | |||
| Disease-free survival | ||||
| T stage (1, 2 vs 3, 4) | < .001* | .018* | 2.188 | 1.145–4.182 |
| N stage (0 vs 1, 2) | < .001* | .269 | 1.386 | 0.777–2.474 |
| Histological grade (1, 2 vs 3) | < .001* | .013* | 1.450 | 1.082–1.942 |
| Lymphovascular invasion (presence vs absence) | < .001* | .041* | 1.873 | 1.027–3.418 |
| MMR (intact vs defective) | .001* | .056 | 1.293 | 0.994–1.683 |
| Smad4/PTEN | ||||
| +/+ vs –/+ or +/– | .079 | .402 | 1.177 | 0.804–1.724 |
| –/+ or +/– vs –/– | .006* | .018* | 1.407 | 1.059–1.868 |
| +/+ vs –/– | < .001* | .007* | 1.715 | 1.155–2.545 |
| Overall survival | ||||
| T stage (1, 2 vs 3, 4) | < .001* | .023 | 2.611 | 1.140–5.980 |
| N stage (0 vs 1, 2) | < .001* | .351 | 1.361 | 0.712–2.601 |
| Histological grade (1, 2 vs 3) | < .001* | .003* | 1.635 | 1.185–2.257 |
| Lymphovascular invasion (presence vs absence) | < .001* | .128 | 1.688 | 0.861–3.308 |
| MMR (intact vs defective) | < .001* | < .001* | 1.712 | 1.267–2.315 |
| Smad4/PTEN | ||||
| +/+ vs –/+ or +/– | .007* | .035* | 1.660 | 1.035–2.661 |
| –/+ or +/– vs –/– | .011* | .048* | 1.373 | 1.003–1.881 |
| +/+ vs –/– | < .001* | .001* | 2.238 | 1.379–3.630 |
- 1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65: 87-108. ArticlePubMedPDF
- 2. Cunningham D, Atkin W, Lenz HJ, et al. Colorectal cancer. Lancet 2010; 375: 1030-47. ArticlePubMed
- 3. Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988; 319: 525-32. ArticlePubMed
- 4. Valcz G, Sipos F, Krenács T, et al. Increase of alpha-SMA(+) and CK (+) cells as an early sign of epithelial-mesenchymal transition during colorectal carcinogenesis. Pathol Oncol Res 2012; 18: 371-6. ArticlePubMedPDF
- 5. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993; 363: 558-61. ArticlePubMedPDF
- 6. Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol 2003; 21: 1174-9. ArticlePubMed
- 7. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010; 138: 87.e3.Article
- 8. Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318: 1108-13. PubMed
- 9. Starr TK, Allaei R, Silverstein KA, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009; 323: 1747-50. ArticlePubMedPMC
- 10. Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene 2006; 25: 7531-7. ArticlePubMedPDF
- 11. Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015; 21: 1350-6. PubMedPMCPDF
- 12. Choi D, Lee HW, Hur KY, et al. Cancer stem cell markers CD133 and CD24 correlate with invasiveness and differentiation in colorectal adenocarcinoma. World J Gastroenterol 2009; 15: 2258-64. ArticlePubMedPMC
- 13. Jing F, Kim HJ, Kim CH, Kim YJ, Lee JH, Kim HR. Colon cancer stem cell markers CD44 and CD133 in patients with colorectal cancer and synchronous hepatic metastases. Int J Oncol 2015; 46: 1582-8. ArticlePubMed
- 14. Sahlberg SH, Spiegelberg D, Glimelius B, Stenerlöw B, Nestor M. Evaluation of cancer stem cell markers CD133, CD44, CD24: association with AKT isoforms and radiation resistance in colon cancer cells. PLoS One 2014; 9: e94621. ArticlePubMedPMC
- 15. Chen KL, Pan F, Jiang H, et al. Highly enriched CD133(+)CD44(+) stem-like cells with CD133(+)CD44(high) metastatic subset in HCT116 colon cancer cells. Clin Exp Metastasis 2011; 28: 751-63. ArticlePubMedPDF
- 16. Kechagioglou P, Papi RM, Provatopoulou X, et al. Tumor suppressor PTEN in breast cancer: heterozygosity, mutations and protein expression. Anticancer Res 2014; 34: 1387-400. PubMed
- 17. Saal LH, Gruvberger-Saal SK, Persson C, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet 2008; 40: 102-7. PubMedPDF
- 18. Wang X, Huang H, Young KH. The PTEN tumor suppressor gene and its role in lymphoma pathogenesis. Aging (Albany NY) 2015; 7: 1032-49. ArticlePubMedPMC
- 19. Zhang LL, Mu GG, Ding QS, et al. Phosphatase and tensin homolog (PTEN) represses colon cancer progression through inhibiting paxillin transcription via PI3K/AKT/NF-kappaB pathway. J Biol Chem 2015; 290: 15018-29. PubMedPMC
- 20. Park SH, Won J, Kim SI, et al. Molecular testing of brain tumor. J Pathol Transl Med 2017; 51: 205-23. ArticlePubMedPMCPDF
- 21. Miyaki M, Kuroki T. Role of Smad4 (DPC4) inactivation in human cancer. Biochem Biophys Res Commun 2003; 306: 799-804. ArticlePubMed
- 22. Xu X, Kobayashi S, Qiao W, et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest 2006; 116: 1843-52. ArticlePubMedPMC
- 23. Chow JY, Cabral JA, Chang J, Carethers JM. TGFbeta modulates PTEN expression independently of SMAD signaling for growth proliferation in colon cancer cells. Cancer Biol Ther 2008; 7: 1694-9. PubMedPMC
- 24. Ding Z, Wu CJ, Chu GC, et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 2011; 470: 269-73. PubMedPMC
- 25. Teng Y, Sun AN, Pan XC, et al. Synergistic function of Smad4 and PTEN in suppressing forestomach squamous cell carcinoma in the mouse. Cancer Res 2006; 66: 6972-81. ArticlePubMedPDF
- 26. Yan P, Klingbiel D, Saridaki Z, et al. Reduced expression of SMAD4 is associated with poor survival in colon cancer. Clin Cancer Res 2016; 22: 3037-47. ArticlePubMedPDF
- 27. Liu J, Cho SN, Akkanti B, et al. ErbB2 pathway activation upon Smad4 loss promotes lung tumor growth and metastasis. Cell Rep 2015; 10: 1599-613. ArticlePubMedPMC
- 28. Xu X, Ehdaie B, Ohara N, Yoshino T, Deng CX. Synergistic action of Smad4 and PTEN in suppressing pancreatic ductal adenocarcinoma formation in mice. Oncogene 2010; 29: 674-86. ArticlePubMedPDF
- 29. Jang KS, Song YS, Jang SH, et al. Clinicopathological significance of nuclear PTEN expression in colorectal adenocarcinoma. Histopathology 2010; 56: 229-39. ArticlePubMed
- 30. Peppercorn J, Perou CM, Carey LA. Molecular subtypes in breast cancer evaluation and management: divide and conquer. Cancer Invest 2008; 26: 1-10. ArticlePubMed
- 31. Stambolic V, Suzuki A, de la Pompa JL, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998; 95: 29-39. ArticlePubMed
- 32. Davies MA, Koul D, Dhesi H, et al. Regulation of Akt/PKB activity, cellular growth, and apoptosis in prostate carcinoma cells by MMAC/PTEN. Cancer Res 1999; 59: 2551-6. PubMed
- 33. Haeger SM, Thompson JJ, Kalra S, et al. Smad4 loss promotes lung cancer formation but increases sensitivity to DNA topoisomerase inhibitors. Oncogene 2016; 35: 577-86. ArticlePubMedPDF
- 34. Ahmed S, Bradshaw AD, Gera S, Dewan MZ, Xu R. The TGF-beta/Smad4 signaling pathway in pancreatic carcinogenesis and its clinical significance. J Clin Med 2017; 6: 5.PubMedPMC
- 35. Tachibana M, Shibakita M, Ohno S, et al. Expression and prognostic significance of PTEN product protein in patients with esophageal squamous cell carcinoma. Cancer 2002; 94: 1955-60. ArticlePubMed
- 36. Lee HS, Lee HK, Kim HS, Yang HK, Kim WH. Tumour suppressor gene expression correlates with gastric cancer prognosis. J Pathol 2003; 200: 39-46. ArticlePubMed
- 37. Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997; 275: 1943-7. ArticlePubMed
- 38. Zhao Y, Zheng R, Li J, Lin F, Liu L. Loss of phosphatase and tensin homolog expression correlates with clinicopathological features of non-small cell lung cancer patients and its impact on survival: a systematic review and meta-analysis. Thorac Cancer 2017; 8: 203-13. PubMedPMC
- 39. Risinger JI, Hayes AK, Berchuck A, Barrett JC. PTEN/MMAC1 mutations in endometrial cancers. Cancer Res 1997; 57: 4736-8. PubMed
- 40. Yang L, Kuang LG, Zheng HC, et al. PTEN encoding product: a marker for tumorigenesis and progression of gastric carcinoma. World J Gastroenterol 2003; 9: 35-9. ArticlePubMedPMC
- 41. Dicuonzo G, Angeletti S, Garcia-Foncillas J, et al. Colorectal carcinomas and PTEN/MMAC1 gene mutations. Clin Cancer Res 2001; 7: 4049-53. PubMed
- 42. Guanti G, Resta N, Simone C, et al. Involvement of PTEN mutations in the genetic pathways of colorectal cancerogenesis. Hum Mol Genet 2000; 9: 283-7. ArticlePubMedPDF
- 43. Reynisdóttir I, Polyak K, Iavarone A, Massagué J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev 1995; 9: 1831-45. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Precision oncolytic viral therapy in colorectal cancer: Genetic targeting and immune modulation for personalized treatment (Review)
Muhammad Haris Sultan, Qi Zhan, Yigang Wang, Yulong Xia, Xiaoyuan Jia
International Journal of Molecular Medicine.2025; 56(1): 1. CrossRef - Focal Adhesion Kinase Intersects With the BRD4‐MYC Axis and YAP1 to Drive Tumor Cell Growth, Phenotypic Plasticity, Stemness, and Metastatic Potential in Colorectal Cancer
Rongbo Han, Junfeng Shi, Kai Cheng, Zian Wang, Yecang Chen, Orion Spellecy, Abu Saleh Mosa Faisal, Isha Aryal, Jinfei Chen, Rolf Craven, Olivier Thibault, Lauren Baldwin, Lawrence D. Brewer, Sonia Erfani, Chi Wang, Zhenheng Guo, Eric Chen, Burton Yang, Fr
Cancer Medicine.2025;[Epub] CrossRef - Association between the expression of epithelial–mesenchymal transition (EMT)-related markers and oncologic outcomes of colorectal cancer
Mona Hany Emile, Sameh Hany Emile, Amr Awad El-Karef, Mohamed Awad Ebrahim, Ibrahim Eldosoky Mohammed, Dina Abdallah Ibrahim
Updates in Surgery.2024; 76(6): 2181. CrossRef - The Potential Role of Genomic Signature in Stage II Relapsed Colorectal Cancer (CRC) Patients: A Mono-Institutional Study
Michela Roberto, Giulia Arrivi, Emanuela Pilozzi, Andrea Montori, Genoveffa Balducci, Paolo Mercantini, Andrea Laghi, Debora Ierinò, Martina Panebianco, Daniele Marinelli, Silverio Tomao, Paolo Marchetti, Federica Mazzuca
Cancer Management and Research.2022; Volume 14: 1353. CrossRef - Alterations of PTEN and SMAD4 methylation in diagnosis of breast cancer: implications of methyl II PCR assay
Menha Swellam, Entsar A. Saad, Shimaa Sabry, Adel Denewer, Camelia Abdel Malak, Amr Abouzid
Journal of Genetic Engineering and Biotechnology.2021; 19(1): 54. CrossRef - E3 ubiquitin ligase HECW1 promotes the metastasis of non-small cell lung cancer cells through mediating the ubiquitination of Smad4
Chen Lu, Guangyao Ning, Panpan Si, Chunsheng Zhang, Wenjian Liu, Wei Ge, Kai Cui, Renquan Zhang, Shenglin Ge
Biochemistry and Cell Biology.2021; 99(5): 675. CrossRef - Computational quantification of global effects induced by mutations and drugs in signaling networks of colorectal cancer cells
Sara Sommariva, Giacomo Caviglia, Silvia Ravera, Francesco Frassoni, Federico Benvenuto, Lorenzo Tortolina, Nicoletta Castagnino, Silvio Parodi, Michele Piana
Scientific Reports.2021;[Epub] CrossRef - Clinicopathological characterization of SMAD4-mutated intestinal adenocarcinomas: A case-control study
Xiaoyan Liao, Yansheng Hao, Xiaofei Zhang, Stephen Ward, Jane Houldsworth, Alexandros D. Polydorides, Noam Harpaz, Aldo Scarpa
PLOS ONE.2019; 14(2): e0212142. CrossRef - Clinicopathological Characterization and Prognostic Implication of SMAD4 Expression in Colorectal Carcinoma
Seung-Yeon Yoo, Ji-Ae Lee, Yunjoo Shin, Nam-Yun Cho, Jeong Mo Bae, Gyeong Hoon Kang
Journal of Pathology and Translational Medicine.2019; 53(5): 289. CrossRef - Dissecting the therapeutic implications of the complex SMAD4 regulatory network in metastatic colorectal cancer
Ion Cristóbal, Blanca Torrejón, Andrea Santos, Melani Luque, Marta Sanz-Alvarez, Federico Rojo, Jesús García-Foncillas
European Journal of Surgical Oncology.2018; 44(8): 1283. CrossRef - Reply to: Dissecting the therapeutic implications of the complex SMAD4 regulatory network in metastatic colorectal cancer
Jordan M. Cloyd, Takashi Mizuno, Jean-Nicolas Vauthey
European Journal of Surgical Oncology.2018; 44(8): 1285. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
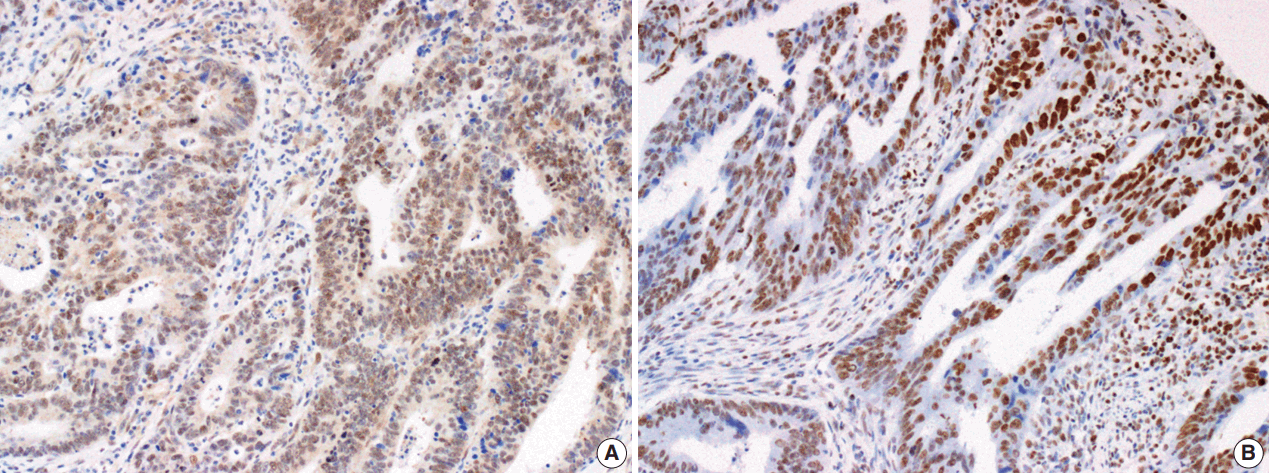
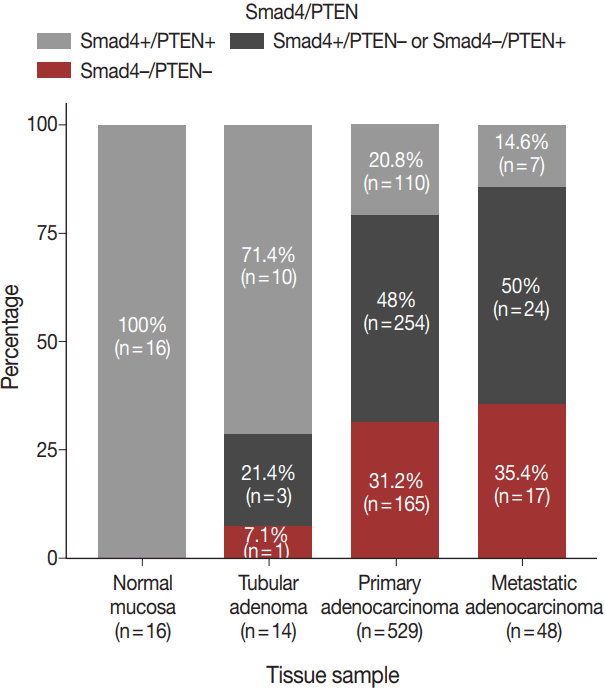
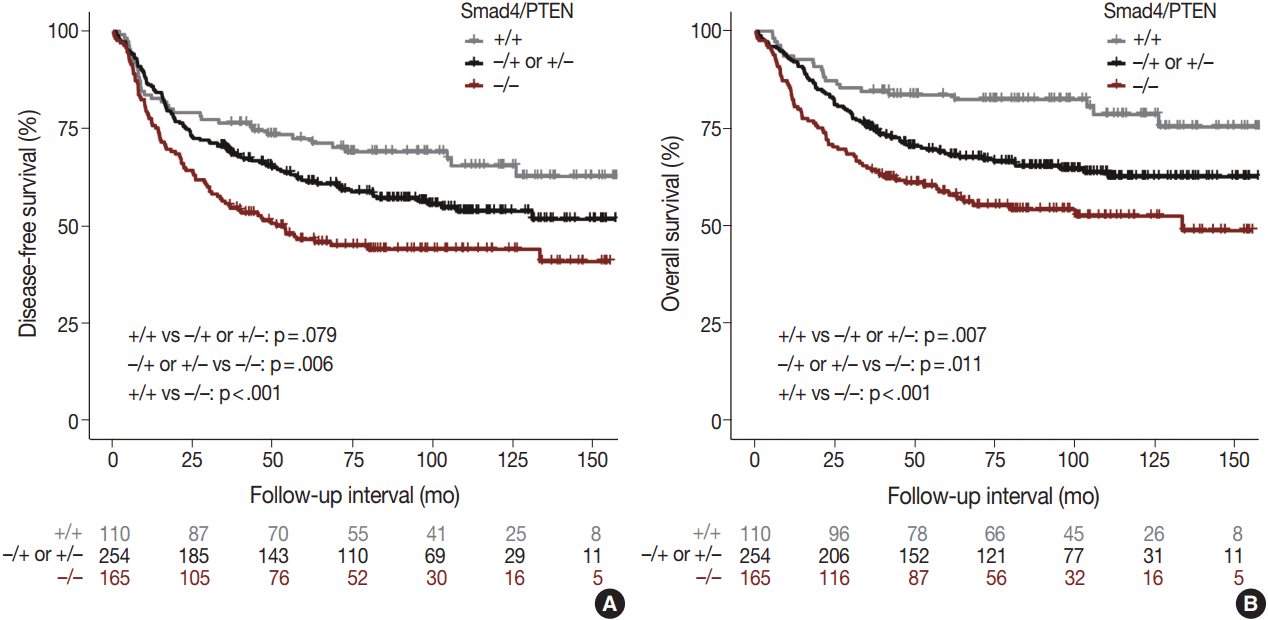



Fig. 1.
Fig. 2.
Fig. 3.
| Parameter | No. (n = 275) | Smad4+/PTEN+ (n = 110) | Smad4–/PTEN– (n = 165) | p-value (chi-square test) |
|---|---|---|---|---|
| Age (yr) | .367 | |||
| < 65 | 189 | 79 (71.8) | 110 (66.7) | |
| ≥ 65 | 86 | 31 (28.2) | 55 (33.3) | |
| Sex | .519 | |||
| Female | 121 | 51 (46.4) | 70 (42.4) | |
| Male | 154 | 59 (53.6) | 95 (57.6) | |
| Tumor location | .413 |
|||
| Right | 63 | 27 (24.5) | 36 (21.8) | |
| Transverse and left | 25 | 12 (10.9) | 13 (7.9) | |
| Rectosigmoid | 187 | 71 (64.5) | 116 (70.3) | |
| Tumor size (cm) | .472 | |||
| ≤ 4.5 | 98 | 42 (38.2) | 56 (33.9) | |
| > 4.5 | 177 | 68 (61.8) | 109 (66.1) | |
| Growth pattern | .866 | |||
| Fungating | 71 | 29 (26.4) | 42 (25.5) | |
| Ulcerating | 204 | 81 (73.6) | 123 (74.5) | |
| Histologic grade | .726 |
|||
| 1 | 13 | 3 (2.7) | 10 (6.1) | |
| 2 | 200 | 83 (75.5) | 117 (70.9) | |
| 3 | 62 | 24 (21.8) | 38 (23) | |
| AJCC stage | .135 |
|||
| Tis | 5 | 1 (0.9) | 4 (2.4) | |
| I | 20 | 12 (10.9) | 8 (4.8) | |
| II | 101 | 44 (40) | 57 (34.5) | |
| III | 149 | 53 (48.2) | 96 (58.2) | |
| T stage | .251 |
|||
| Tis | 5 | 1 (0.9) | 4 (2.4) | |
| 1 | 007 | 5 (4.5) | 2 (1.2) | |
| 2 | 22 | 12 (10.9) | 10 (6.1) | |
| 3 | 231 | 89 (80.9) | 142 (86.1) | |
| 4 | 10 | 3 (2.7) | 7 (4.2) | |
| N stage | .018 |
|||
| 0 | 126 | 57 (51.8) | 69 (41.8) | |
| 1 | 67 | 30 (27.3) | 37 (22.4) | |
| 2 | 82 | 26 (35.8) | 59 (35.8) | |
| Lymphovascular invasion | .138 | |||
| Absence | 125 | 56 (50.9) | 69 (41.8) | |
| Presence | 150 | 54 (49.1) | 96 (58.2) | |
| Mismatch repair protein | .006 |
|||
| Intact | 147 | 70 (63.6) | 77 (46.7) | |
| Defective | 128 | 40 (36.4) | 88 (53.3) |
| Univariate significance | Multivariate significance | HR | 95% CI | |
|---|---|---|---|---|
| p-value |
p-value |
|||
| Disease-free survival | ||||
| T stage (1, 2 vs 3, 4) | < .001 |
.018 |
2.188 | 1.145–4.182 |
| N stage (0 vs 1, 2) | < .001 |
.269 | 1.386 | 0.777–2.474 |
| Histological grade (1, 2 vs 3) | < .001 |
.013 |
1.450 | 1.082–1.942 |
| Lymphovascular invasion (presence vs absence) | < .001 |
.041 |
1.873 | 1.027–3.418 |
| MMR (intact vs defective) | .001 |
.056 | 1.293 | 0.994–1.683 |
| Smad4/PTEN | ||||
| +/+ vs –/+ or +/– | .079 | .402 | 1.177 | 0.804–1.724 |
| –/+ or +/– vs –/– | .006 |
.018 |
1.407 | 1.059–1.868 |
| +/+ vs –/– | < .001 |
.007 |
1.715 | 1.155–2.545 |
| Overall survival | ||||
| T stage (1, 2 vs 3, 4) | < .001 |
.023 | 2.611 | 1.140–5.980 |
| N stage (0 vs 1, 2) | < .001 |
.351 | 1.361 | 0.712–2.601 |
| Histological grade (1, 2 vs 3) | < .001 |
.003 |
1.635 | 1.185–2.257 |
| Lymphovascular invasion (presence vs absence) | < .001 |
.128 | 1.688 | 0.861–3.308 |
| MMR (intact vs defective) | < .001 |
< .001 |
1.712 | 1.267–2.315 |
| Smad4/PTEN | ||||
| +/+ vs –/+ or +/– | .007 |
.035 |
1.660 | 1.035–2.661 |
| –/+ or +/– vs –/– | .011 |
.048 |
1.373 | 1.003–1.881 |
| +/+ vs –/– | < .001 |
.001 |
2.238 | 1.379–3.630 |
Values are presented as number (%). AJCC, 8th edition of American Joint Committee on Cancer. p < .05. Linear by linear association.
HR, hazard ratio; CI, confidence interval; MMR, mismatch repair protein. p < .05. Log rank test; Cox proportional hazard model adjusted for T and N stage, histological grade, lymphovascular invasion, MMR and Smad4/PTEN expression.

