The Smad4/PTEN Expression Pattern Predicts Clinical Outcomes in Colorectal Adenocarcinoma
Article information
Abstract
Background
Smad4 and PTEN are prognostic indicators for various tumor types. Smad4 regulates tumor suppression, whereas PTEN inhibits cell proliferation. We analyzed and compared the performance of Smad4 and PTEN for predicting the prognosis of patients with colorectal adenocarcinoma.
Methods
Combined expression patterns based on Smad4+/– and PTEN+/– status were evaluated by immunostaining using a tissue microarray of colorectal adenocarcinoma. The relationships between the protein expression and clinicopathological variables were analyzed.
Results
Smad4–/PTEN– status was most frequently observed in metastatic adenocarcinoma, followed by primary adenocarcinoma and tubular adenoma (p<.001). When Smad4–/PTEN– and Smad4+/PTEN+ groups were compared, Smad4–/PTEN– status was associated with high N stage (p=.018) and defective mismatch repair proteins (p=.006). Significant differences in diseasefree survival and overall survival were observed among the three groups (Smad4+/PTEN+, Smad4–/PTEN+ or Smad4+/PTEN–, and Smad4–/PTEN–) (all p<.05).
Conclusions
Concurrent loss of Smad4 and PTEN may lead to more aggressive disease and poor prognosis in patients with colorectal adenocarcinoma compared to the loss of Smad4 or PTEN alone.
Colorectal cancer (CRC) is a heterogeneous disease with diverse clinical presentations, responses to therapy, and histopathological findings at diagnosis [1,2]. The development of CRC is linked to homeostatic balance through the regulation of cellular proliferation, differentiation, and migration [3,4]. Approximately 15% of all CRCs have microsatellite instability (MSI) as a consequence of a deficient mismatch repair (MMR) system involving proteins such as MLH1, MSH2, and MSH6 [5-7]. CRC with MSI is associated with various clinicopathological features, including a tendency to arise in the proximal colon, lymphatic invasion, high histological grade, and chemotherapeutic resistance.
Recent molecular studies have demonstrated several genetic and epigenetic alterations in CRC [8-10]. The four consensus molecular subtypes of CRC are CMS1 (MSI immune), CMS2 (canonical, marked WNT and MYC signaling activation), CMS3 (metabolic dysregulation), and CMS4 (mesenchymal, prominent transforming growth factor β [TGF-β] activation, stromal invasion, and angiogenesis) [11]. High expression of CD133, CD44, and CD24 in cancer stem cells has been shown to be correlated with worse clinicopathological features in CRC [12,13]. Moreover, high coexpression of CD133 and CD44 has been associated with the AKT pathway and increased radiation resistance in colon cancer cells [14,15].
Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) is a phosphoinositide 3-phosphatase that can inhibit cellular proliferation, survival, growth, and differentiation in several types of neoplasm such as colon, breast, and lymphoid cell neoplasms [16-20]. Smad4 (DPC4) is a tumor suppressor gene that mediates the TGF-β signaling pathway, thus suppressing epithelial cell growth [21]. Loss of Smad4 is linked to cancers arising in different organs such as the pancreas, colon, prostate, and bile duct, and it is also linked to gastric polyposis [21-26]. Ablation of Smad4 and PTEN in the pulmonary epithelium has been shown to induce metastatic adenosquamous carcinoma via the ErbB2/ELF3/AKT signaling pathway [27] and one in vivo study showed that coordinated deletion of the Smad4 and PTEN genes accelerated skin and stomach carcinogenesis [25]. In addition, PTEN deficiency has been shown to initiate widespread precancerous lesions triggered by Smad4 deficiency [28].
Based on these data, we hypothesized that the loss of both Smad4 and PTEN is significantly associated with more aggressive CRC behavior than the loss of either protein individually. The purpose of this study was to investigate the expression patterns of Smad4 and PTEN in patients with CRC. We evaluated whether the combination of Smad4 and PTEN expression showed better performance for predicting patient survival than either marker alone.
MATERIALS AND METHODS
Patients and tissue specimens
This retrospective study enrolled a consecutive series of 529 patients with colorectal adenocarcinoma, 48 patients with metastatic adenocarcinoma, 14 patients with tubular adenoma, and 16 patients with normal colonic mucosae. All patients were diagnosed and treated at our institute from January 1991 to August 2001. This study included data obtained from a previously conducted research study [15]. The Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK) criteria were followed throughout this study. Inclusion criteria included histopathological evidence of adenocarcinoma confirmed by a pathologist and known clinical outcome. Exclusion criteria included diagnosis of adenocarcinoma with neoadjuvant chemotherapy or inadequate clinical history. Clinicopathological findings were collected from the patients’ medical records and pathological reports. Patient age ranged from 17 to 87 years (mean, 57.7 years). The tumors were located at the cecum (n = 18), ascending colon (n = 78), hepatic flexure (n = 12), transverse colon (n=26), splenic flexure (n = 4), descending colon (n = 25), sigmoid colon (n = 113), and rectum (n = 253). Tumor size ranged from 3 to 150 mm (mean, 57 mm). A mean number of 26.7 lymph nodes (LN) were dissected for each specimen. Over a mean follow-up interval after surgery of 5.9 years, 183 patients (34.6%) died and 346 (65.4%) survived. Normal colonic mucosae, tubular adenomas, and metastatic adenocarcinoma tissues were randomly selected for evaluation of Smad4 and PTEN expression.
This study (involving human participants) was approved by the Ethics Committee of Hanyang University Hospital (No. 2016-12-030-001) and was performed in accordance with the ethical standards of the Declaration of Helsinki, as revised in 2008. The institutional review board review confirmed that informed consent was not necessary for this study.
Tissue microarray construction
A representative area was carefully selected and marked on a hematoxylin and eosin (H&E)-stained slide. Tissue microarrays (TMAs) were assembled using a tissue-array instrument (Quick-Ray Manual Tissue Microarrayer, Unitma Co. Ltd., Seoul, Korea) consisting of thin-walled stainless steel punches and stylets used to empty and transfer the needle content. Areas rich in tumor cells were identified by light microscopic examination of H&Estained sections and selected for use in TMAs. Tissue cylinders with a diameter of 2 mm were punched from the previously marked tumor area (tumor center) of each donor block and transferred to a recipient paraffin block. All tissue cores were composed of tumor tissue in more than 70% of the core area. Considering the limitations associated with obtaining representative areas of a tumor, we used a 2-mm tissue core from each donor block.
Immunohistochemical staining and scoring of Smad4 and PTEN expression
Primary mouse monoclonal anti-Smad4 antibodies (clone B-8, Santa Cruz Biotechnology Inc, Heidelberg, Germany) were diluted 1:250 and primary mouse monoclonal anti-PTEN antibodies (clone 28H6, Novocastra Laboratories, Newcastle upon Tyne, UK) were diluted 1:200.
The Smad4 and PTEN immunohistochemical (IHC) staining results were interpreted by two pathologists (K.J. and S.S.P.) who were blinded to the clinical data. Discordant cases were reviewed on a multi-headed microscope to reach consensus. Each slide was interpreted semi-quantitatively according to the intensity of nuclear immunoreactivity (Fig. 1). The cut-off was determined by proportion rather than by intensity, because the latter was rather heterogeneous. Smad4 and PTEN expression was classified as negative (< 10% positive tumor cells out of all cancer cells) or positive (≥ 10% positive tumor cells out of all cancer cells) [29].
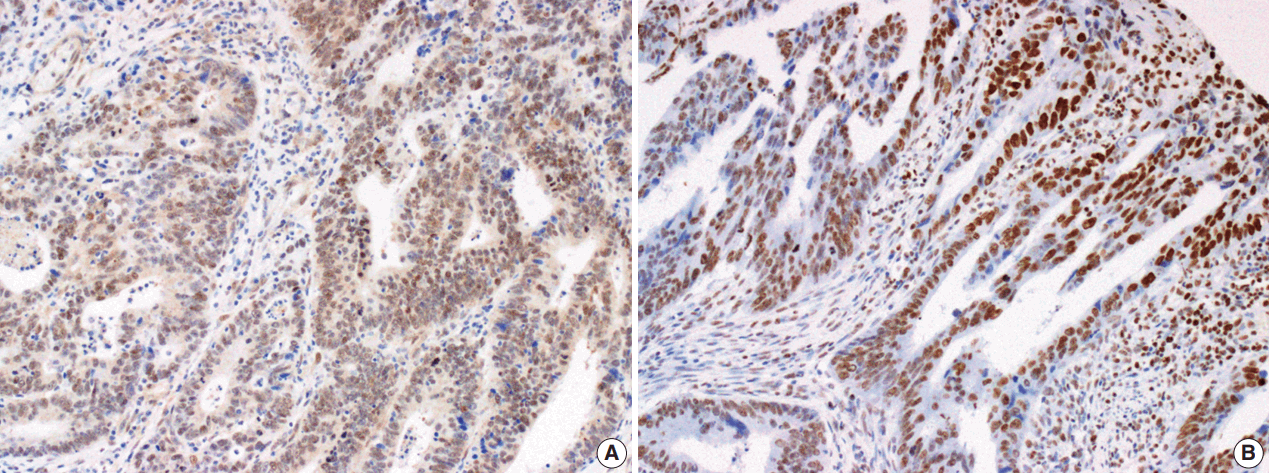
Microphotographs showing immunohistochemical staining of Smad4 (A) and PTEN (B) expression in colorectal adenocarcinoma tissue.
Statistical analysis
The chi-square test was used to evaluate potential associations between Smad4 expression/PTEN expression and clinicopathologic parameters including age, gender, tumor location, tumor size, growth pattern, histological grade, of American Joint Committee on Cancer (AJCC) stage (T and N criteria), and MMR protein expression. The relationships between Smad4 and PTEN expression and normal mucosae, adenomatous polyps, adenocarcinomas, and metastatic LNs were analyzed using linear-by-linear association. Overall survival (OS) was defined as the time from curative surgery to death from any cause. Diseasefree survival (DFS) was defined as the time from curative surgery to the first tumor recurrence or distant metastasis. Kaplan–Meier curves with the log-rank test were used to calculate OS and DFS. Multivariate survival analysis was used with a Cox proportional hazards regression model to evaluate independent prognostic factors. A p-value of < .05 was considered significant. Statistical analysis was performed using SPSS ver. 20.0 (IBM Corp., Armonk, NY, USA) or R packages (http://www.r-project.org/).
RESULTS
The expression patterns of Smad4 and PTEN were evaluated in normal colonic mucosae, tubular adenomas, primary adenocarcinomas, and metastatic adenocarcinomas. The loss of both Smad4 and PTEN was associated with cancer progression. Specifically, Smad4–/PTEN– status was not observed in any of the 16 normal colonic mucosa tissues; however, it was observed in one of 14 tubular adenomas (7.1%), 165 of 529 primary adenocarcinomas (31.2%), and 17 of 48 metastatic adenocarcinomas (35.4%) (p < .001) (Fig. 2).
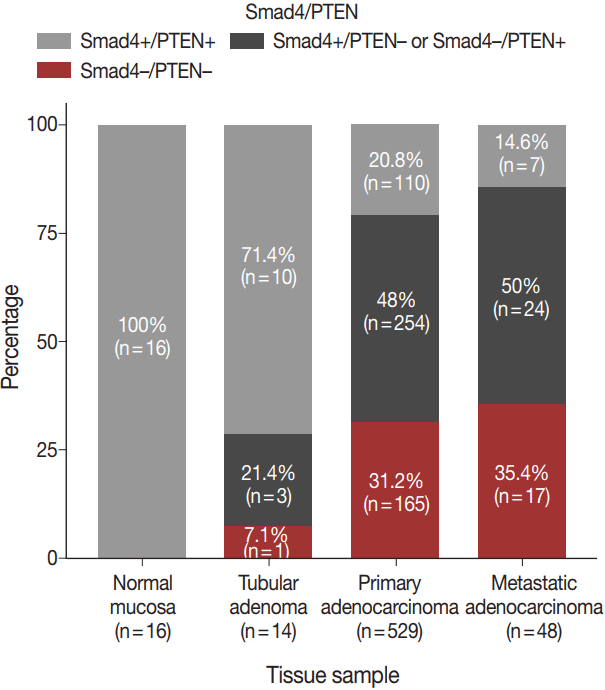
Proportions of different tissues showing Smad4–/PTEN–staining: normal (0%), adenoma (7.2%), primary adenocarcinoma (31.2%), and metastatic adenocarcinoma (35.4%).
Smad4 loss was associated with higher N stage (p = .013) and showed a tendency to correlate with lymphovascular invasion (p = .081) (Supplementary Table S1). PTEN loss was correlated with defective MMR (p = .005) (Supplementary Table S2).
The distribution of Smad4 and PTEN expression status was as follows: Smad4–/PTEN–, 165 patients; Smad4–/PTEN+, 155 patients; Smad4+/PTEN–, 99 patients; and Smad4+/PTEN+, 110 patients.
Comparison of the Smad4–/PTEN– and Smad4+/PTEN+ groups showed that Smad4–/PTEN– status was associated with higher N stage (p = .018) and defective MMR (p = .006) (Table 1). In a comparative analysis between the Smad4+/PTEN+ and Smad4–/PTEN+ or Smad4+/PTEN– groups, Smad4+/PTEN+ status was correlated with lower N stage (p = .047), absence of lymphovascular invasion (p = .049), and intact MMR (p = .006) (Supplementary Table S3). Another comparative analysis between the Smad4–/PTEN+ or Smad4+/PTEN– and Smad4–/PTEN– groups showed that Smad4–/PTEN– status was correlated with defective MMR (p = .030) (Supplementary Table S4).

Correlations between clinicopathological parameters and Smad4/PTEN in 275 patients with colon cancer
Survival according to Smad4/PTEN expression
Univariate and multivariate survival analyses showed that the loss of Smad4 was associated with short DFS and OS (all p < .05). In univariate analyses, loss of PTEN was not associated with short DFS or OS (all p < .05). In multivariate analyses, no relationship was observed between DFS or OS and PTEN.
Comparison of the Smad4+/PTEN+ and Smad4–/PTEN– groups showed that Smad4–/PTEN– status was associated with short DFS and OS in both univariate and multivariate analyses (all p < .05). Comparison of the Smad4–/PTEN+ or Smad4+/PTEN– and Smad4–/PTEN– groups revealed significant differences in DFS and OS (all p <. 05). Comparison of the Smad4+/PTEN+ and Smad4–/PTEN+ or Smad4+/PTEN– groups showed a significant difference in OS (p = .035), whereas the difference in DFS was not significant (Table 2, Fig. 3).

Disease-free and overall survival analyses according to expression patterns of Smad4/PTEN
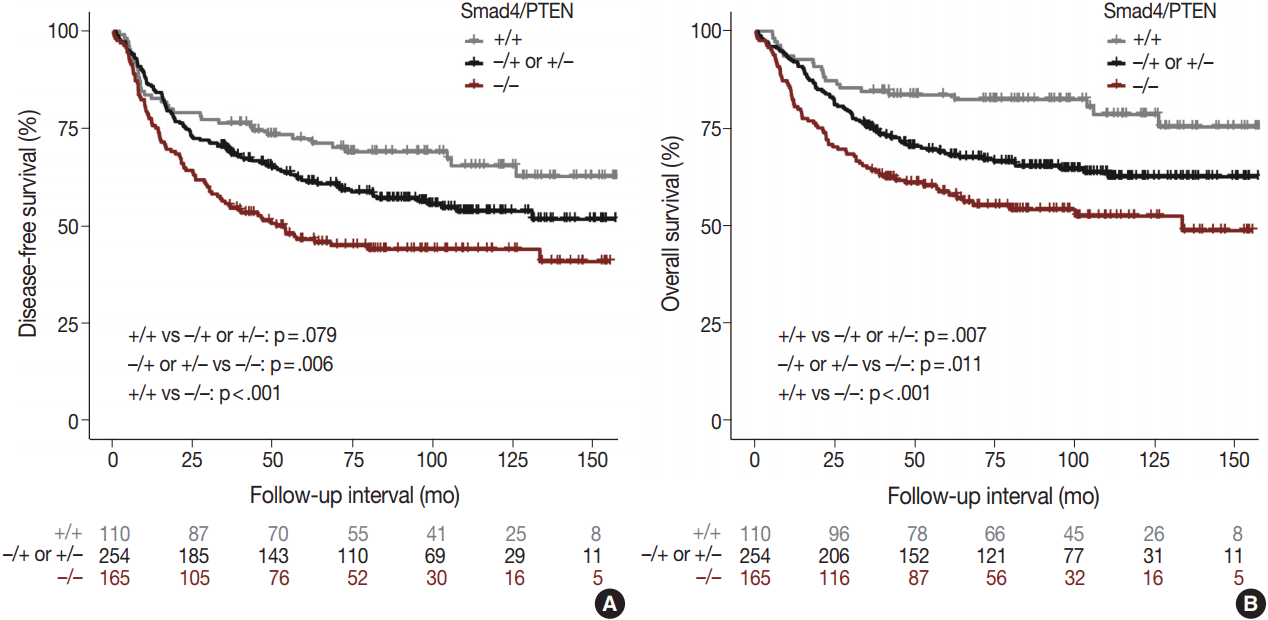
Disease-free survival (A) and overall survival (B) curves derived by the Kaplan–Meier method showing the correlation of survival with Smad4/PTEN expression status.
DISCUSSION
The aim of this study was to determine whether the Smad4/PTEN expression pattern is associated with clinical outcomes. Smad4 is part of a protein complex linked to signal transduction and interacts with DNA for cell proliferation [30]. PTEN is a negative regulator of the Akt signaling pathway and is necessary for cell proliferation and inhibition of apoptosis [31,32]. Smad4 and PTEN have been shown to be closely linked in the regulation of tumor invasion and distant metastasis. For instance, Haeger et al. [33] demonstrated that the loss of Smad4 could initiate lung cancer development from keratinized epithelium in vivo. Moreover, loss of Smad4 expression and low Smad4 expression were both shown to increase cell proliferation secondary to the loss of TGF-β– mediated growth suppression [34]. During cancer development, deletion of PTEN is common in several malignancies such as gastric, esophageal, endometrial, breast, and non-small cell lung cancers [25,35-40]. Moreover, many studies have identified PTEN mutations in HRS320 cells, a colonic cancer cell line, and shown that these mutations are significantly related to poor clinical outcomes in patients with CRC [41,42].
The prognostic value of combinations of cancer markers for patients with CRC (which exhibits high heterogeneity) may be better than that of models using a single marker. An in vivo study demonstrated that loss of both Smad4 and PTEN may act synergistically to regulate cell proliferation and accelerate tumorigenesis [25]. The synergistic action of these genes is mediated by concomitant repression of PTEN transcription by Smad4 and promotion of Smad4 degradation by PTEN [22]. Liu et al. [27] showed that the combined loss of Smad4 and PTEN was linked to metastatic lung cancer through activation of the ErbB2/ELF3/AKT pathway. In pancreatic cancer, the absence of Smad4 and PTEN has been shown to synergistically promote ductal carcinoma [28]. Intriguingly, Smad4 deletion has been shown to drive progression of PTEN-deficient prostate cancer to highly aggressive behaviors such as tumor invasion and lymph node metastasis [24]. Thus, Smad4 deficiency can accelerate tumor cell proliferation and invasion, especially when combined with loss of PTEN expression. Although this relationship has been confirmed in an in vitro study, no reports have described a difference in DFS or OS in human CRC according to the loss of both Smad4 and PTEN. In the present study, we showed that Smad4+/PTEN+ status indicated good prognosis, whereas Smad4−/PTEN– status was associated with poor prognosis. These findings suggest that the loss of both Smad4 and PTEN expression is linked to worse clinical outcomes in patients with CRC. In addition, Smad4–/PTEN– status was significantly associated with higher N stage compared to Smad4+/PTEN+ status.
The precise mechanism underlying the synergistic effect between Smad4 and PTEN is not fully understood. In esophageal cancer, it has been proposed that the loss of both Smad4 and PTEN leads to the downregulation of CIP/KIP and INK4 Cdk inhibitors and subsequent suppression of cell cycle arrest [43]. Alternatively, concurrent deletion of PTEN and Smad4 has been reported to induce an oncogenic pathway including ErbB2/Akt/ELF3 and to repress tumor suppressors in lung cancer [27]. Interestingly, the lung cancer study demonstrated that the mRNA level of ELF3 (a transcriptional factor known to have oncogenic activity) was significantly higher in Smad4–/PTEN– mice compared to Smad4– mice and PTEN– mice. In pancreatic cancer, Smad4–/PTEN– status was associated with wider cancer spread and larger tumors compared with Smad4– status or PTEN– status [28]. In the present study, the Smad4–/PTEN+ and Smad4+/PTEN– groups had lower survival rates than the Smad4+/PTEN+ group, but higher survival rates than the Smad4–/PTEN– group. These biological processes, together with the concurrent loss of Smad4 and PTEN, may correlate with more aggressive clinical behavior than that seen with the loss of a single protein.
This study did have some limitations. First, in contrast to previous in vitro studies, we did not demonstrate a molecular interaction between Smad4 and PTEN, making it difficult to draw concrete conclusions from our data. Second, the cut-off value of 10% for distinguishing positive from negative expression of Smad4 and PTEN was determined by IHC evaluation. However, Smad4 and PTEN distribution may vary according to specimen type, implying that this cut-off point may not apply to other types of clinical samples or to other laboratories. Third, interactions between Smad4 and PTEN within single tumor cells could not be observed, because double staining of Smad4 and PTEN expression was not performed.
In summary, we found that the Smad4/PTEN expression pattern was statistically correlated with several clinicopathological parameters and with survival. Smad4–/PTEN– status was more frequently observed in patients who followed the multi-step carcinogenesis progression from normal colonic mucosae to tubular adenoma, primary adenocarcinoma, and metastatic adenocarcinoma. Some differences in survival were observed among the Smad4+/PTEN+, Smad4+/PTEN– or Smad4–/PTEN+, and Smad4–/PTEN– groups. Thus, combinations of markers may be more useful for predicting the survival of patients with CRC than single markers alone. We conclude that concurrent loss of Smad4 and PTEN may accelerate cancer progression and that these two genes thus represent potential targets for cancer treatment.
Electronic Supplementary Material
Supplementary materials are available at Journal of Pathology and Translational Medicine (http://jpatholtm.org).
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.