Molecular Screening of Small Biopsy Samples Using Next-Generation Sequencing in Korean Patients with Advanced Non-small Cell Lung Cancer: Korean Lung Cancer Consortium (KLCC-13-01)
Article information
Abstract
Background
Non-small cell lung cancer (NSCLC) is a common type of cancer with poor prognosis. As individual cancers exhibit unique mutation patterns, identifying and characterizing gene mutations in NSCLC might help predict patient outcomes and guide treatment. The aim of this study was to evaluate the clinical adequacy of molecular testing using next-generation sequencing (NGS) for small biopsy samples and characterize the mutational landscape of Korean patients with advanced NSCLC.
Methods
DNA was extracted from small biopsy samples of 162 patients with advanced NSCLC. Targeted NGS of genomic alterations was conducted using Ion AmpliSeq Cancer Hotspot Panel v2.
Results
The median age of patients was 64 years (range, 32 to 83 years) and the majority had stage IV NSCLC at the time of cancer diagnosis (90%). Among the 162 patients, 161 patients (99.4%) had novel or hotspot mutations (range, 1 to 21 mutated genes). Mutations were found in 41 genes. Three of the most frequently mutated genes were TP53 (151, 93.2%), KDR (104, 64.2%), and epidermal growth factor receptor (EGFR; 69, 42.6%). We also observed coexistence of EGFR and other oncogene (such as KRAS, PIC3CA, PTEN, and STK11) mutations. Given that 69.6% (48/69) of EGFR mutant patients were treated with EGFR tyrosine kinase inhibitors, EGFR mutant status had higher prognostic ability in this study.
Conclusions
These results suggest that targeted NGS using small biopsy samples is feasible and allows for the detection of both common and rare mutations in NSCLC.
The majority of patients diagnosed with non-small cell lung cancer (NSCLC) present with advanced stage disease and have extremely poor prognosis [1]. Recent advances in the understanding of lung cancer biology and improvements in technology have allowed molecular classification of NSCLC [2,3]. Classifying NSCLC into distinct actionable subtypes with mutually exclusive driver oncogenes has led to the development of targeted therapy [3]. Better survival has been observed after treatment with epidermal growth factor receptor (EGFR) or anaplastic lymphoma kinase (ALK) tyrosine kinase inhibitors (TKIs) in patients harboring the appropriate activating mutation or translocation, compared with standard chemotherapy [3,4].
Although several genes including EGFR and ALK have been identified as potential oncogenic drivers and targets for therapy, a large fraction of NSCLC patients do not have mutations in these commonly mutated genes. Thus, there are needs to identify additional driver oncogenes and targets for treatment. In addition, many NSCLC patients also harbor other co-existing molecular alterations that might influence the efficacy of a targeted therapy, leading to primary or secondary resistance. It is important to investigate these concurrent genetic alterations to reveal clinically significant predictive and prognostic markers. However, several challenges remain in the implementation of multiple molecular tests to find therapeutic or prognostic markers. First, most NSCLC biopsy samples are not amenable to multiple molecular tests due to the small amounts of tissues obtained by bronchoscopy or core biopsy. In addition, conventional molecular tests such as Sanger sequencing and polymerase chain reaction (PCR) are insensitive to alterations occurring at allele frequencies lower than 20%. Finally, multiple and separate tests result in higher costs and longer turn-around time. Thus, a more comprehensive, sensitive, and time/cost-effective multiplex test is necessary to optimize the application of targeted therapy [5,6]. Consequently, incorporation of molecular screening using nextgeneration sequencing (NGS) in the pathologic evaluation of NSCLC is now considered the standard in clinical practice [7,8].
The rapid development of NGS technologies has enabled a new paradigm in precision medicine for oncology. It is now possible to identify oncogenic alterations that would have been previously undiscovered by conventional tests such as sequencing. For the routine clinical molecular diagnostic testing in NSCLC, NGS need to meet some criteria; NGS platform should be able to detect targetable driver mutations from limited amounts of input DNA from small biopsy or cytology samples, the turn-around time should be short, and the cost should be low. Unlike whole-genome sequencing or whole-exome sequencing, targeted NGS including selected genes that show frequent alterations in cancer can reduce the amount of tissue, time, and cost required for testing [9-11].
To validate the accuracy and feasibility of targeted NGS, we used Ion AmpliSeq Cancer Hotspot Panel v2 to identify the variety of tumor-associated mutations in formalin-fixed paraffin-embedded (FFPE) or fresh frozen (FF) specimens from 162 advanced NSCLC patients in Korea. In this study, we analyzed multiple somatic mutations found in our advanced NSCLC cohort in order to detect known actionable mutations and discover potential therapeutic targets and prognostic biomarkers for NSCLC.
MATERIALS AND METHODS
Patients and tumor samples
We analyzed 162 FFPE or frozen tumor tissue specimens from advanced NSCLC patients between January 2014 and December 2015 at Samsung Medical Center (SMC). All samples were collected before any treatments were initiated. Procedures used for tumor tissue sampling varied, including video-assisted thoracoscopic surgery, core-needle biopsy, bronchoscopy, and endobronchial ultrasonography. Clinical data were obtained retrospectively from electronic medical records. The clinical variables assessed were sex, age at diagnosis, smoking history, tumor subtype, cancer stage, EGFR mutation, ALK rearrangement, chemotherapy regimen, TKIs, and tumor response. Separately, EGFR mutation status was tested by real-time PCR using the peptide nucleic acid (PNA)–clamping EGFR Mutation Detection Kit (Panagene, Inc., Daejeon, Korea). Real-time PCR was performed using a CFX96 (Bio-Rad, Hercules, CA, USA) and all reagents were included in the kit. PCR cycling and mutation detection were done as previously described [12]. ALK rearrangement status was tested by immunohistochemistry and confirmed by fluorescence in situ hybridization (FISH). All procedures involving tumor specimens were reviewed and approved by the Institutional Review Board (IRB) of SMC and all data were fully anonymized (SMC 2013-08-113-020). Written informed consent was provided by all patients.
DNA extraction
All tissue sections were reviewed by pathologists, and only those with tumor content more than 10% were included in the study. Genomic DNA was extracted from FFPE samples using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) or FF tumor specimens using QIAamp DNA mini kit. Purified DNA was quantitated by NanoDrop (Invitrogen Life Technologies, Carlsbad, CA, USA) and Qubit Fluorometer (Invitrogen Life Technologies).
Next-generation sequencing and data analysis
The Ion Torrent Ion AmpliSeq Cancer Hotspot Panel v2 (Life Technologies) was used. This panel detects hotspot regions, including ~2,800 COSMIC mutations of 50 oncogenes and tumor suppressor genes. A total of 162 cases of NSCLC specimens were subjected to NGS on the AmpliSeq platform. Sequencings were done according to previously described methods [13]. Variants calls were further processed to reduce potential false-positives. Coverage (> 500 ×) was considered as filtering criteria and the minimal variant allele frequency was 2% for confirming variants as real. After filtering using these criteria, variants causing amino acid change and frameshift were finally used for statistical analysis.
Statistical analysis
Clinical and radiological response to treatment was assessed according to Response Evaluation Criteria In Solid Tumor ver. 1.1. Kaplan-Meier estimates were used for the analysis of all time-to-event variables. Progression-free survival (PFS) was calculated from the date of chemotherapy to the date of disease progression or death from any cause or the date of last follow-up. The overall survival (OS) was measured from the date of chemotherapy to the date of death from any cause and was censored at the date of the last follow-up visit. Variables with p < .05 were considered significant. All statistical analyses were performed using PASW Statistics ver. 23.0 (SPSS Inc., Chicago, IL, USA).
RESULTS
Patient characteristics
The clinical characteristics of advanced NSCLC patients included in the present study are summarized in Table 1. The median age was 64 years (range, 32 to 83 years) and gender proportions were roughly equal (male 57% vs female 43%). Seventy-nine patients (59%) were smokers or former smokers, 83 patients (51%) were never smokers. The NSCLC subtype distribution was as follows: adenocarcinoma (139/162, 85.8%), squamous cell carcinoma (17/162, 10.5%), adenosquamous cell carcinoma (1/162, 0.6%), and other (5/162, 3.1%). The majority of patients had stage IV NSCLC at the time of cancer diagnosis (145/162; 90%). In stage IV NSCLC patients, the median PFS was 6.2 months (95% CI, 4.2 to 8.1) and OS was 19.6 months (95% CI, 15.4 to 23.7). EGFR mutation test was done in 145 patients, and the mutation was detected in 64 patients (44.1%) by realtime PCR using PNA-clamping. Positive results for ALK rearrangement by FISH were detected in 14 patients (8.6%). In this cohort, 81 patients were treated with cytotoxic chemotherapy (81/162, 50.0%), 51 with EGFR TKIs (51/162, 31.5%), and three with ALK TKIs (3/162, 1.8%) as first-line therapy.

The baseline characteristics of patients
Molecular profiling of advanced NSCLC
We employed targeted NGS technology to evaluate somatic mutations occurring in advanced NSCLC, using the Ion Torrent Ion AmpliSeq Cancer Hotspot Panel. Among the detected mutations, only those annotated in the Catalogue of Somatic Mutations in Cancer (COSMIC) database were considered. Mutations were found in 41 genes and commonly detected in the following genes: TP53 (151, 93.2%), KDR (104, 64.2%), EGFR (69, 42.6%), APC (51, 31.5%), RB1 (30, 18.5%), SMAD4 (28, 17.3%), MET (22, 13.6%), STK11 (20, 12.3%), RET (18, 11.1%), ALK (17, 10.5%), and KRAS (13, 8.0%), as shown in Fig. 1. Only one patient had no mutation while 161 (99.4%) patients possessed more than one mutation (range, 1 to 21; median, 4). The vast majority of identified mutations were single nucleotide variant (SNV) followed by deletion (Del) and insertion (Ins). In accordance with the frequency described in previous studies [1,3], EGFR mutations were found in 42.6% patients and most of them (54/69, 78.3%) were typical mutations (30 Del exon 19 and 24 L858R). One of these was a triple EGFR mutant (L858R/G873R/Q787L) and ten were double EGFR mutant (3 Del exon 19/G873R, 2 Del exon 19/A750P, 1 Del exon 19/S752Y, 1 Del exon 19/K754N, 2 L858R/G873R, 1 L858R/T790M). Besides TP53 and EGFR, the most frequently mutated gene was KDR, and KDR mutations appeared in codon 472 (103 Q472H) and codon 875 (1 T875A). One patient with KDR Q472H had concurrent KDR S1148C. MET mutations were found in codon 375 (17 N375S) and codon 179 (5 A179T). Although one MET R970C in exon 14 was simultaneously found with N375S, this has not been reported as cause of MET exon 14 skipping [14]. STK11 mutations were found in codon 354 (19 F354L) and codon 176 (1 D176G). Two samples detecting STK11 F354L had other STK11 mutation in codon 281 (P281L). Seventeen ALK mutations were all in codon 1184 (G1184E). In concordance with the known frequency of KRAS mutations in Asian population (5%–10%) [15], they were found in 8% of this cohort. Most KRAS mutations appeared in codon 12 (1 G12A, 4 G12C, 3 G12D, 1 G12R, and 2 G12V) with two mutations in codon 50 (T50P). PIK3CA mutations (E81K, R401Q, E542G, E545A, E545K, Q546K, and H1047R) were detected in seven samples and one patient had double PIK3CA mutants (E542G/E545A). PTEN mutations (K66E, R130X, Q171X, and P246L) were identified in four patients.
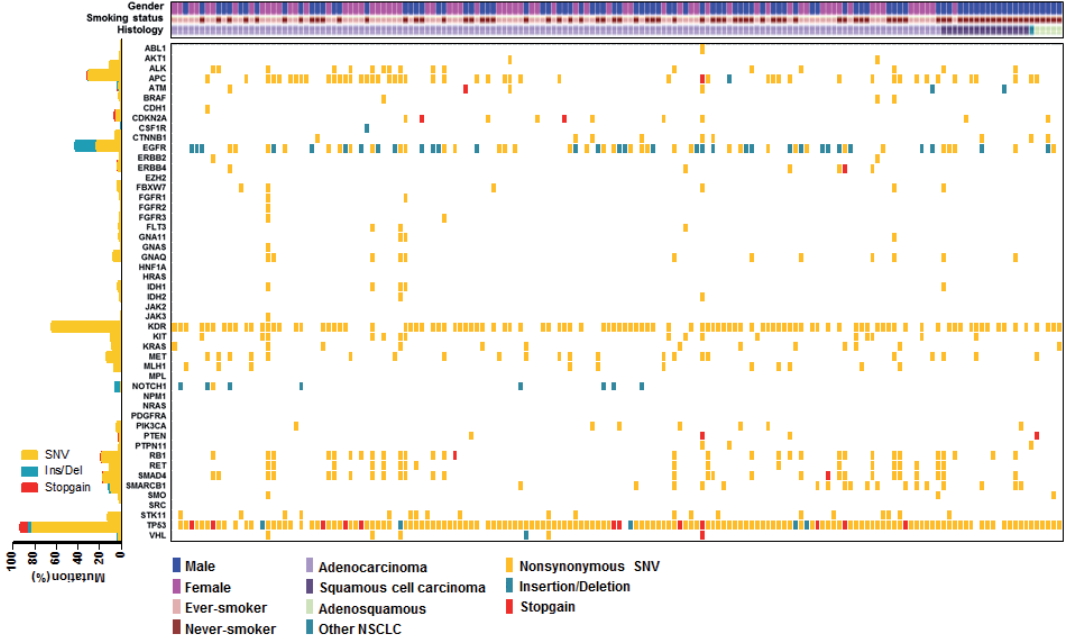
Heatmap of mutations found in 162 non-small cell lung cancer samples. In the upper panel, the first row indicates sex, the second row smoking status, and the third row histology. A histogram shows the percentage of mutations in each gene (let). The horizontal axis presents the complete dataset of patients and the vertical axis illustrates mutated genes (right).
We also observed co-occurrence of some of the most frequently mutated and clinically significant genes. Five patients simultaneously had mutations in both EGFR and KRAS. EGFR mutations also harbored PIK3CA and PTEN mutations, which were detected in three and two patients, respectively. In addition, although STK11 mutations were most commonly seen in association with KRAS mutations, we found seven cases with co-occurrence of EGFR and STK11 mutations in this study.
Comparison of mutational profiles obtained with the AmpliSeq assay
Based on mutation results considering the location of mutation sites, EGFR mutations were consistently detected by targeted NGS using AmpliSeq Cancer Panel and conventional PNA-clamping PCR (42.6% vs 44.1%). In 145 patients tested for EGFR mutation, the comparison results of EGFR mutations detected by targeted NGS and conventional PNA-clamping PCR are summarized in Table 2. When comparing mutation detection of EGFR in FFPE and FF samples, a high concordance rate (92.4%) was seen between NGS and PNA-clamping PCR. However, targeted NGS method identified additional EGFR mutations in 14 concordant cases and seven discordant cases that were not identified by PNA-clamping PCR. The most frequently found additional EGFR mutation was G873R which was found in 12 patients. We observed three discordant cases that showed positive results (all Del exon 19) in PNA-clamping, but negative in NGS. Considering the high sensitivity of NGS, these results may be due to tumor heterogeneity.
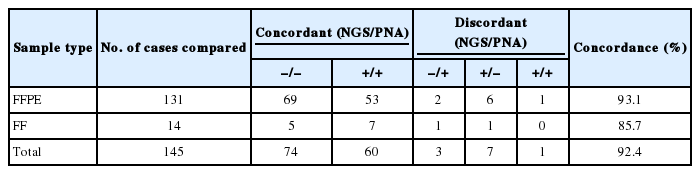
Comparison of EGFR mutations detected by targeted NGS and PNA-clamping PCR
Although most of the patients had a single biopsy, four patients had repeated biopsies and had double tumors tested. Except for one consistent case, the other three cases showed slightly different mutation profiles (Table 3). These differences may be due to tumor heterogeneity or tumor evolution.
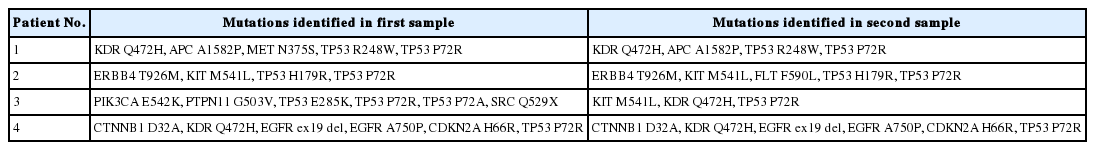
Mutations identified in four patients with repeat biopsy samples
Impact of mutation status on survival
We evaluated the relationships between EGFR somatic mutations and survival. Activating EGFR mutations have been reported as prognostic factors in other studies [16,17]. In this study cohort, 69.6% (48/69) of patients with EGFR mutations were treated with EGFR TKIs. The presence of EGFR mutations were definitive predictive markers of both PFS (hazard ratio [HR], 2.59; 95% confidence interval [CI], 1.75 to 3.85) (Fig. 2A) and OS (HR, 2.00; 95% CI, 1.30 to 3.09) (Fig. 2B). Median PFSs were 3.8 months for EGFR wild-type group and 14.6 months for EGFR mutant group. The median OS for EGFR wild-type group was 14.8 months, but the median OS for EGFR mutant group was not reached. When PFS was analyzed after grouping the patients according to the three types of EGFR mutations (Del ex19, L858R, and others), the median PFSs were different between activating mutations (15.1 months for Del ex19 and 17.7 months for L858R) and others (6.6 months) (Fig. 3A). However, the median OS was not reached in the three types of EGFR mutations (Fig. 3B).
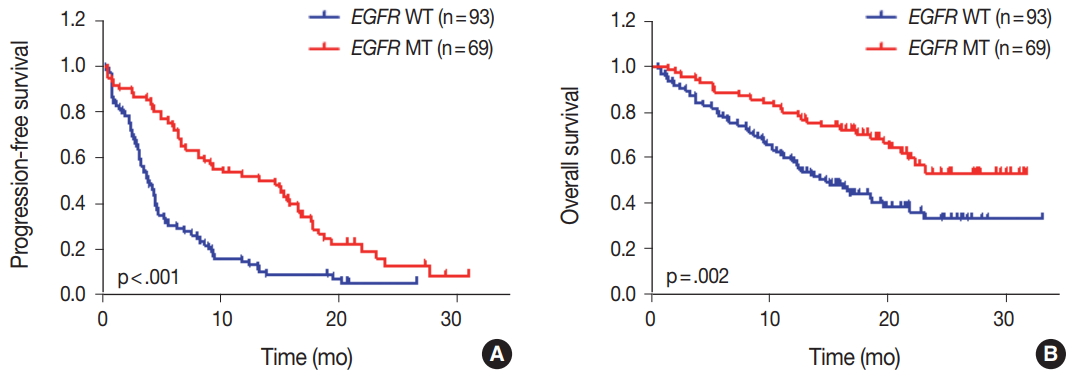
Impact of epidermal growth factor receptor (EGFR) mutational status on survival. (A) Progression-free survival of patients with EGFR mutant (MT) compared with EGFR wild-type (WT) patients. (B) Overall survival of all patients according to EGFR status. p-values were obtained using the log-rank test.
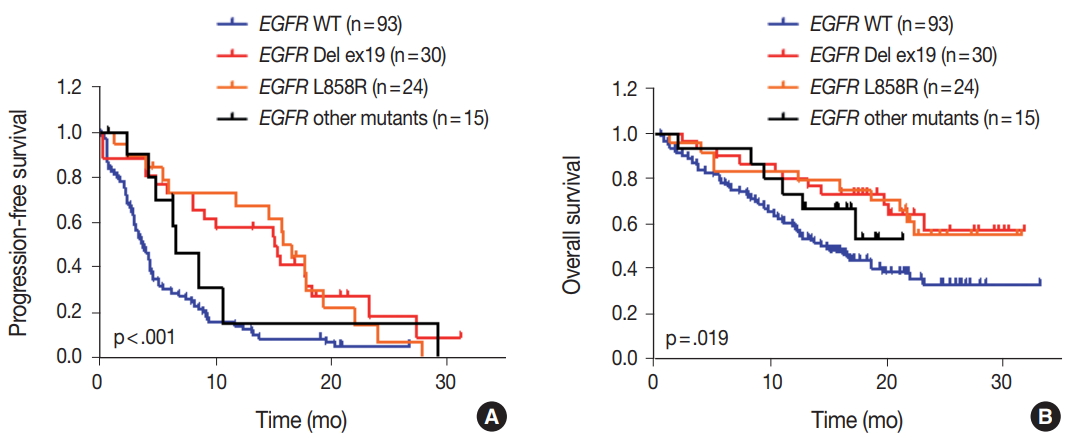
Impact of different types of epidermal growth factor receptor (EGFR) mutations on survival. Progression-free survival (A) and overall survival (B) of patients with different types of EGFR mutations compared with EGFR wild type (WT) patients. p-values were obtained using the log-rank (Mantel-Cox) test.
DISCUSSION
Currently, molecular testing for EGFR mutations and ALK rearrangements is essential for targeted therapy in patients with NSCLC. However, the heterogeneous nature of NSCLC can lead to inaccurate molecular classification and therapeutic resistance. Other genetic alterations that have been found and have potential therapeutics include ROS1, RET, and NTRX gene rearrangement, MET exon 14 skipping, and MET amplification [4,18,19]. However, there are no standard molecular diagnostic tests for these genetic alterations. In addition, an important limitation in current routine diagnosis is that the quantity of DNA extracted from small biopsy samples (FFPE or FF) is not adequate for multiple molecular tests in most cases. To cope with this limitation, comprehensive multiplex testing using NGS is necessary to improve the efficacy of targeted therapy for NSCLC patients. With advances in NGS technology, several target regions of interest can be sequenced concurrently and thereby improve the chances of identifying rare mutations. In this study, 162 Korean advanced NSCLC samples were assessed for mutations in oncogenes and tumor suppressor genes using an NGS platform (AmpliSeq Cancer Hotspot Panel). This targeted sequencing method shows high accuracy and requires only small quantities of sample (10 ng DNA), enabling researchers to sequence challenging small biopsy samples such as FFPE. Genetic alterations were confirmed in 99.4% of samples and 14 additional EGFR mutations (L707F, G719A, G719C, L747S, A750P, S752Y, K754N, S768I, V769L, V774M, T783A, S784P, Q787L, and G873R) were identified that were not detected with PNA-clamping PCR. In addition, we found some of the most frequently altered and clinically significant genes such as KRAS, MET, STK11, PIK3CA, and PTEN mutations. Moreover, the higher sensitivity of NGS platform should increase the identification of concomitant mutations. These results suggest the feasibility and usefulness of targeted sequencing to identify low frequency mutations and detect additional mutations that are helpful to understand the clinical outcomes of the patients in each group.
For patients with EGFR-mutant NSCLC, EGFR TKIs are found to increase response rates and survival time [16,17]. In concordance with these data, EGFR mutations were associated with significant improvements of PFS and OS compared to EGFR wild-type patients, because most patients were treated with EGFR TKIs. Despite these benefits of EGFR TKIs, not all patients respond to treatment and most EGFR-mutant NSCLC patients develop acquired resistance.
Tumor suppressor TP53 mutations are frequently detected in most human cancers. TP53 was also the most commonly altered gene in this study, and this result is consistent with those of previous studies [20,21]. TP53 was concurrently mutated with many other genes such as EGFR and KRAS in this study, perhaps due to the high frequency of TP53 mutations found in our samples. Whereas the frequency of TP53 mutation is well known, therapeutic options based on this alteration are scarce and controversial in patients with lung cancer. A previous study on advanced NSCLC found an association between TP53 mutation and shorter median OS, but another study, on the other hand, reported no association between TP53 mutation and survival [22,23].
Our data also identified KDR Q472H polymorphism in 103 patients (31 homozygotes and 72 heterozygotes). KDR Q472H has been reported to increase tumor microvasculature and shown to mediate vascular endothelial growth factor receptor 2 phosphorylation in NSCLC [24]. Furthermore, KDR Q472H had a higher proliferative and invasive capacity in melanoma [25]. Although we did not find a significant correlation between KDR Q472H and survival in EGFR TKI- or chemotherapytreated NSCLC patients, the prognostic value of KDR Q472H should be different after treatment with vascular endothelial growth factor pathway inhibitors.
Although KRAS mutations are the most common oncogenic driver, there are some ethnic differences. The frequency of KRAS mutations in Asian is 5%–15%. In addition, KRAS mutations usually occur in EGFR wild-type tumors [18,26]. In this study, we detected 13 KRAS mutations (8%) and five concurrent KRAS/EGFR mutations (3.0%) via NGS. In these five patients, three patients, who were clinically confirmed to have EGFR L858R mutations, received EGFR TKI (gefitinib) treatment with partial response or progression of the disease. Other two patients were treated with chemotherapy and showed 0.8 and 9.3 months of PFS. Although the prognostic effect of KRAS mutations was not clear due to small sample size, these results suggest that KRAS mutation test using NGS platform may help determine the appropriate therapy for NSCLC patients.
Recent studies have demonstrated that mutations in EGFR-downstream genes such as PIK3CA, PTEN, and STK11 are associated with de novo resistance to EGFR TKI [27]. Furthermore, PIK3CA and PTEN mutations may result in resistance to EGFR TKI [4]. In this study, we found PIK3CA and PTEN mutations in seven (4.3%) and four (2.5%) patients, respectively. Concurrent EGFR/PIK3CA mutations were detected in three patients. All of them received EGFR TKI (2 gefitinib and 1 erlotinib) treatment and showed partial response with different range of PFS (6.6–21.3 months). Concurrent EGFR/PTEN mutations were found in two patients and one received EGFR TKI (afatinib) treatment with partial response (PFS, 8.1 months). However, neither PIK3CA nor PTEN mutation status alone had significant effects on PFS and OS in the EGFR-mutant group. In STK11, we identified mutations in 20 patients (19 F354L and 1 D176G). STK11 encodes the serine/threonine protein kinase and is part of the STK11/AMPK/mammalian target of rapamycin signaling pathway. STK11 mutations were commonly found, and inactivation of STK11 is known to promote tumorigenesis and is associated with worse survival outcome [20,28]. The overall rate of STK11 mutations (12.3%) was slightly lower than that indicated by The Cancer Genome Atlas (17%) [18]. This discrepancy can be explained by the origin of the population; STK11 mutations have been reported to be associated with European ancestry [19,21]. STK11 mutations often coexist with KRAS mutations and have confounding prognostic significance [29,30]. However, in this study, we found only one concurrent KRAS/STK11 (G12A/D176G) mutation. This patient received chemotherapy (AP: doxorubicin, cisplatin) and showed partial response with 4.4 months of PFS. A recent study reported that pathogenic STK11 F354L mutations had been recurrently identified in three EGFR TKI non-responders, while these mutations had not been found in EGFR TKI responders [20]. In our study, seven STK11 F354L mutations were recurrently found in EGRK TKI–treated patients. Among them, six (treated with gefitinib and erlotinib) showed partial response (PFS, 4.1 to 17.8 months) and one (treated with afatinib) showed stable disease (PFS, 19.3 months). This discrepancy may be due to the small sample size of STK11 mutant patients. Thus, more research is required to identify the clinical implications of STK11 mutations.
Our study has a few limitations. Our analysis relied on targeted sequencing to investigate genetic alterations and thus the genes selected in this study may only explain a portion of the total genetic alterations. The NGS platform used in this study (AmpliSeq Cancer Hotspot Panel) detected only SNVs, and thus it was impossible to detect copy number variations (CNVs) and translocations. Furthermore, other novel genetic or epigenetic alterations may have been missed. Most tumor samples were acquired from small biopsy samples, and thus there were not enough tissue available for more comprehensive analysis. Therefore, there is a need for new NGS platforms to simultaneously detect SNVs, CNVs, and translocations, even with small amounts of tissue samples. In addition, functional effects of the detected mutations were not evaluated in vitro.
Our results demonstrate that targeted sequencing using NGS is feasible for mutation profiling of small biopsy samples in NSCLC. We also demonstrated previously unappreciated mutations, enabling further refinements of subclassification for the prediction of therapeutic effects. In conclusion, we suggest that more comprehensive genomic characterizations of NSCLC with small biopsy samples would reveal coexisting alterations that might influence the efficacy of therapy.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI16C1984).