Primary pulmonary epithelioid inflammatory myofibroblastic sarcoma: a rare entity and a literature review
Article information

Abstract
Epithelioid inflammatory myofibroblastic sarcoma (EIMS) is an aggressive subtype of inflammatory myofibroblastic tumor (IMT) harboring anaplastic lymphoma kinase (ALK) gene fusions and is associated with high risk of local recurrence and poor prognosis. Herein, we present a young, non-smoking male who presented with complaints of cough and dyspnoea and was found to harbor a large right lower lobe lung mass. Biopsy showed a high-grade epithelioid to rhabdoid tumor with ALK and desmin protein expression. The patient initially received 5 cycles of crizotinib and remained stable for 1 year; however, he then developed multiple bony metastases, for which complete surgical resection was performed. Histopathology confirmed the diagnosis of EIMS, with ALK gene rearrangement demonstrated by fluorescence in situ hybridization. Postoperatively, the patient is asymptomatic with stable metastatic disease on crizotinib and has been started on palliative radiotherapy. EIMS is a very rare subtype of IMT that needs to be included in the differential diagnosis of ALKexpressing lung malignancies in young adults.
Inflammatory myofibroblastic tumor (IMT) is a distinctive mesenchymal tumor composed of neoplastic myofibroblastic tumor cells associated with inflammatory infiltrate of plasma cells, lymphocytes, and occasional eosinophils or neutrophils. IMTs present mainly in children and young adults, lack significant gender bias, and occur in a variety of anatomical locations [1]; lungs are one of the most common sites, accounting for ~1% of all adults and ~50% of pediatric lung tumors [2]. Fusions of the anaplastic lymphoma kinase (ALK) gene with various fusions partners, most commonly tropomyosin 3 (TPM3) or tropomyosin 4 (TPM4) gene, are observed in up to 60% of IMTs, correlating with diffuse cytoplasmic staining for ALK protein in tumor cells. Conventional IMTs show low-grade spindle cell tumor morphology lacking significant atypia, mitoses, or necrosis. Surgical resection is curative, and although one-fifth of cases will recur, < 5% of all IMTs metastasize [1,2].
Epithelioid inflammatory myofibroblastic sarcoma (EIMS) is a very rare subtype of IMT that has been associated with an aggressive course, rapid local recurrence, early metastases, and fatal outcome [1,3]. Unlike the bland spindle cell morphology of IMT, EIMS is composed of plump round to epithelioid tumor cells with prominent nucleoli and myxoid stroma with neutrophil-rich infiltrate [1] and show mitoses and necrosis. They harbor ALK gene fusions, most commonly with RANBP2 (Ran specific binding protein 2) or, rarely, with RRBP1 (ribosome binding protein 1). The fusions have been associated with a peculiar nuclear membrane (RANBP2-ALK fusion) or perinuclear accentuated cytoplasmic immunostaining (RRBP1-ALK fusion) for ALK protein [3-5]. EIMS occurs mainly in children and young adults, with a male gender predilection. These tumors have been most commonly reported in the mesentery and omentum [3,6], while primary pulmonary EIMS is extremely rare [7-9]. Herein, we report a primary pulmonary EIMS in a young male who was initially treated with ALK inhibitor monotherapy and subsequent surgery in the face of metastatic progression. In addition, we discuss the challenges associated with the diagnosis and treatment of this rare entity.
CASE REPORT
A 25-year-old, non-smoking male presented with complaints of cough, significant weight loss, intermittent high-grade fever, and difficulty breathing for longer than 4 months. Imaging revealed a well-defined homogenous fluorodeoxyglucose (FDG)-avid mass of 6.9 × 4.2 × 6.1 cm involving the superior segment of the right lower lung lobe with no other pulmonary/pleural lesions or lymphadenopathy (Fig. 1A). Positron emission tomography scan showed another FDG-avid well-defined hypodense lesion of size 1.1 × 1.2 cm in segment IVA of the liver, likely metastasis. Biochemistry including serum tumor biomarkers was within normal range. A computed tomography–guided biopsy of the lung mass showed a poorly differentiated malignant tumor immunonegative for epithelial (pan-cytokeratin, epithelial membrane antigen), hematopoietic (CD45, myeloperoxidase, CD30), pneumocytic (thyroid transcription factor 1), melanocytic (human melanoma black 45, S-100), and mesothelial (calretinin, Wilms tumor protein 1) markers with retained INI-1 and BRG-1 expression. Tumor cells showed cytoplasmic ALK (VENTANA anti-ALK [D5F3] antibody developed by Roche Diagnostics [one 5 mL dispenser of VENTANA anti-ALK (D5F3) antibody contains approximately 70 μg of the rabbit monoclonal (D5F3) antibody; the antibody is diluted in 0.08 M phosphate buffered saline with 3% carrier protein and 0.05% Pro-Clin 300, a preservative]) and cytoplasmic desmin. Myogenin was negative. The possibility of EIMS was suggested, and resection/ re-biopsy was advised for molecular confirmation due to limited remaining biopsy material.
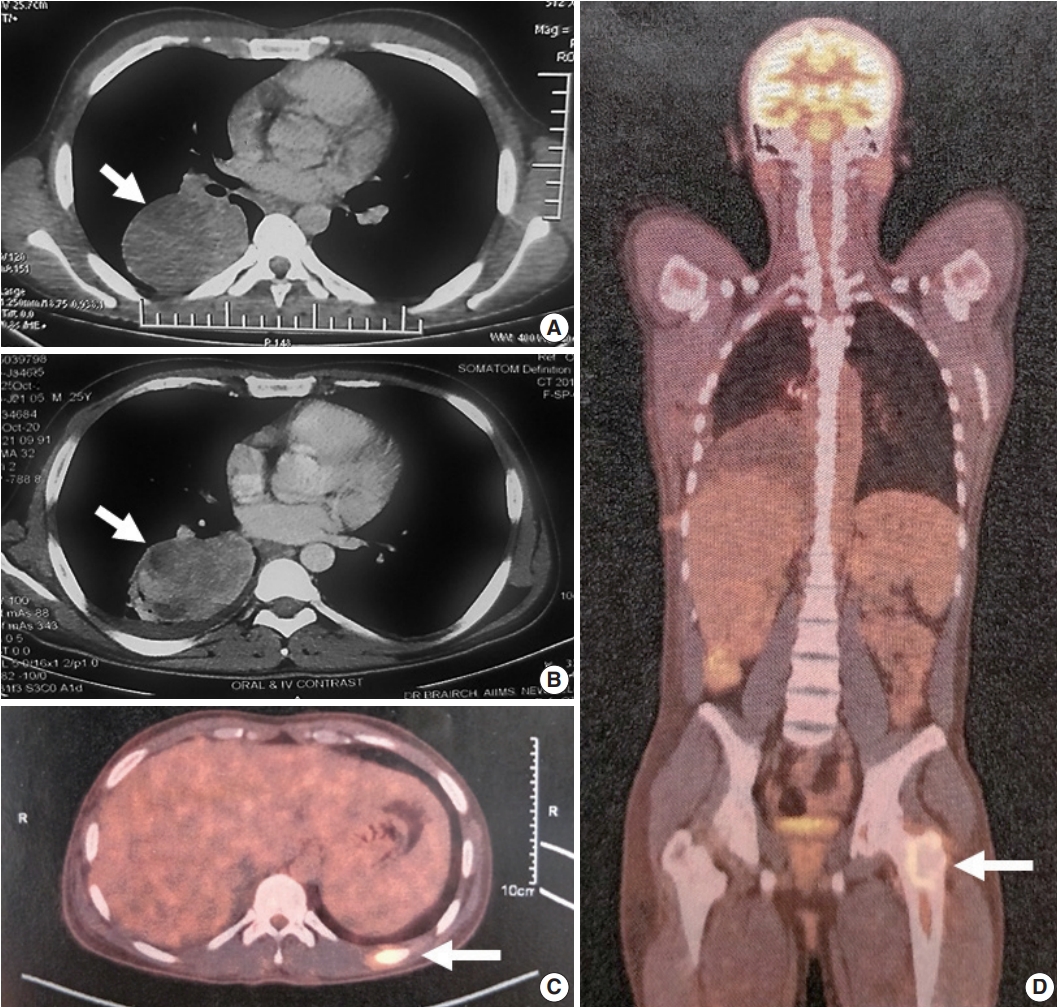
Imaging features of the patient. (A) Computed tomography images show a homogeneous mass (arrow) with well-defined margin involving the superior segment of the right lower lobe. (B) Imaging at 6-months post-biopsy with the patient on crizotinib; the mass (arrow) was stable in size with few hypodense areas suggestive of necrosis. Positron emission tomography scan at 12-month post-biopsy with the patient continuing on crizotinib revealed new metastatic lesions in left 10th rib (arrow, C) and left proximal femur (arrow, D).
After multidisciplinary thoracic oncology tumor board discussion, the patient was planned for surgical excision of the lung tumor and radiofrequency ablation of the liver lesion because of the young age of the patient with good performance status, solitary metastasis, and unconfirmed primary pathological diagnosis. However, the patient was started on ALK inhibitor (crizotinib), and surgical intervention was deferred due to coronavirus disease 2019. Six months later, repeat imaging showed stable pulmonary disease (Fig. 1B) and complete resolution of the liver lesion. Crizotinib was continued and the patient was re-assessed for definitive surgical management. Repeat imaging 1 year after presentation showed increase in the size of the pulmonary mass (7.2 × 5.3 × 4.2 cm), and bone scan revealed metastasis in the right humerus (mid-shaft), left 10th rib, and left proximal femur (Fig. 1C, D). Pleural effusion or mediastinal lymph node metastasis (endobronchial ultrasound-guided transbronchial needle aspiration showed reactive lymphoid hyperplasia) was absent. Despite new bony metastases, because of the young age of the patient, surgical intervention was carried out as planned by means of video-assisted thoracoscopy-assisted right lower lobectomy.
Grossly, a well circumscribed, soft, fleshy tumor was identified in the lung parenchyma with focal areas of haemorrhage and necrosis (Fig. 2). Microscopy (Fig. 3) revealed a homogeneous tumor composed of ovoid to polygonal cells arranged in fascicles and sheets. The cells showed moderate to abundant eosinophilic cytoplasm with markedly pleomorphic nuclei, prominent nucleoli, and fine granular cytoplasm showing frequent mitoses (8 per 2 mm2). Necrosis was present that had not been seen in the biopsy. Focal areas of myxoid change and lymphoplasmacytic infiltrate were noted. The immuno-profile of the tumor was similar to that of the biopsy except for focal cytokeratin expression in the resection, while histomorphology of the tumor was similar to that of the biopsy. The Ki-67 labeling index was ~30%–35% in the highest labeled areas. There was diffuse strong cytoplasmic staining for ALK protein. Fluorescence in situ hybridization assay using the FlexISH ALK/ROS1 DistinguISH Probe (ZytoVision, GmbH, Bremerhaven, Germany) showed one or more isolated 3' signals (extra orange signals) of the ALK gene without corresponding 5' signals (green signals) in the majority of tumor cells, indicative of ALK gene rearrangements [10], confirming the diagnosis of primary pulmonary EIMS. The ROS1 gene represented by fused aqua-green-orange signals was intact (Fig. 3). The postoperative period was unremarkable, and the patient is currently asymptomatic 4-month post-surgery. He is continuing on crizotinib and is planned for palliative radiotherapy for the bony metastases.
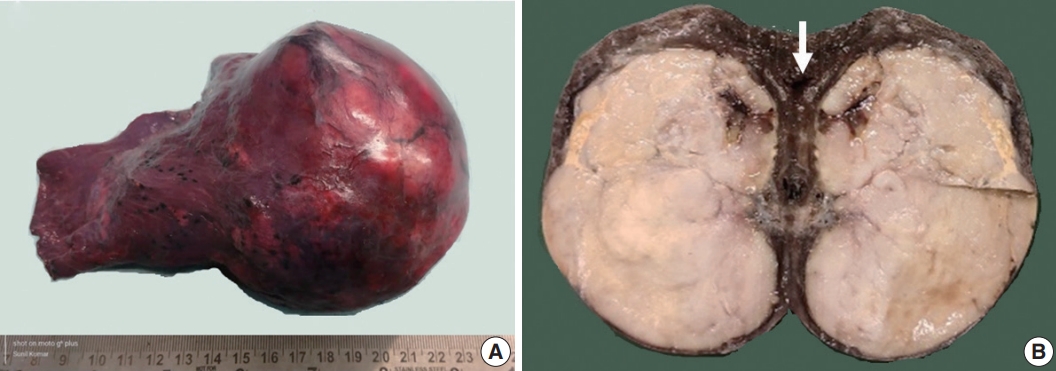
Gross features of the resected pulmonary tumor. Resection specimen shows a well encapsulated, lobulated, uncut tumor (A). A cut section shows a circumscribed, fleshy tumor with a surrounding rim of lung parenchyma (arrow, B).
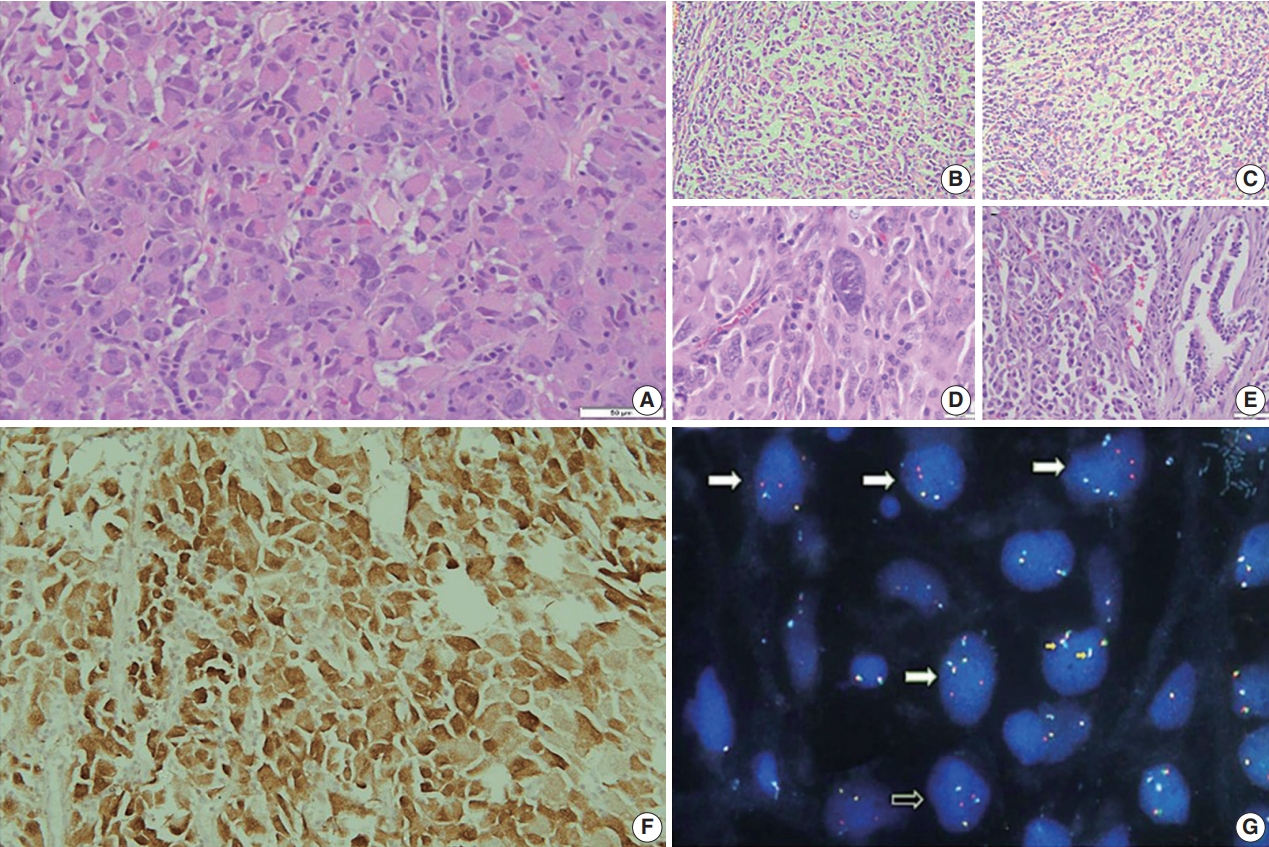
Histomorphological and molecular features of the tumor. Hematoxylin and eosin-stained sections show rhabdoid phenotype tumor cells with moderate to abundant inclusion-like eosinophilic cytoplasm, prominent nucleoli, and fine granular chromatin with frequent mitoses in a myxoid stroma (A).The tumor cells show rich myxoid stroma and infiltration of mixed inflammatory cells (B, C, E) and prominent nuclear pleomorphism (D). The tumor cells show strong and diffuse cytoplasmic anaplastic lymphoma kinase (ALK) immunopositivity (F). (G) Fluorescence in situ hybridization using the ALK-ROS1 FLEXISH probe shows extra 3' signals (spectrum orange) of the ALK gene in tumor cells (white arrows, extra red signals unfused with green and/or aqua), indicating ALK gene rearrangement. The ROS1 gene (indicated by fused red green-aqua signals, small yellow arrows) is intact.
DISCUSSION
Primary pulmonary EIMS is extremely rare, with only three cases reported previously in the English literature to the best of our knowledge (Table 1) [7-9]. Including the present case, primary pulmonary EIMS occurs across a wide age range (21–71 years at diagnosis) with a male preponderance. The tumor presents as a solitary parenchymal mass or as pleural mass with a relatively short symptom duration (< 6 months) and is not uncommonly metastatic at presentation, with brain and skeleton appearing to be the preferred sites of metastases (Table 1).
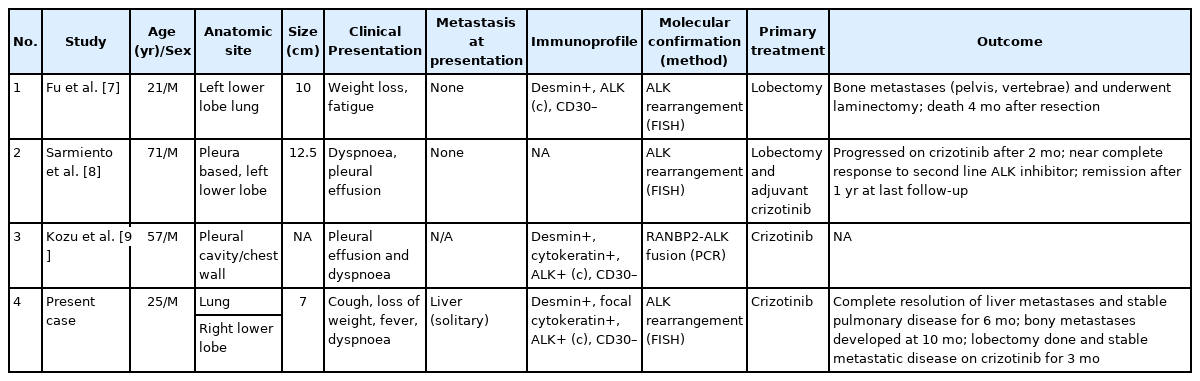
Clinicopathological features of reported cases of primary pulmonary epithelioid inflammatory myofibroblastic sarcoma including the current case
Diagnosis of EIMS is challenging. The poorly differentiated epithelioid morphology can mimic anaplastic large cell lymphoma (ALCL), malignant melanoma, alveolar rhabdomyosarcoma, alveolar soft part sarcoma, epithelioid leiomyosarcoma, undifferentiated sarcoma, malignant mesothelioma, poorly differentiated carcinoma, and pulmonary pleomorphic carcinoma (containing a minimum 10% spindle and/or giant cells) and needs to be differentiated by relevant immunohistochemical markers (Table 2). Conventional IMTs are more common in the lung but are differentiated from EIMS by their bland spindle morphology, prominent lymphoplasmacytic cellular infiltrate, and absence of mitoses and pleomorphism. Rare cases of otherwise conventional IMTs that show increased mitotic activity, necrosis, and aggressive biological behavior are reported [11,12]; these should not be designated as EIMS in the absence of distinctive morphology of round to epithelioid cells, myxoid stroma, and mixed inflammatory infiltrate, as was observed in the present case. ALK protein expression is essential for diagnosis but is not specific as many other lung tumors express ALK protein due to underlying ALK gene rearrangements, as in ALCL, conventional IMTs, and non-small cell lung carcinoma (NSCLC), or to ALK copy number alterations as in rhabdomyosarcomas [13]. ALK-rearranged lung adenocarcinoma/NSCLC shows a variety of histological characteristics ranging from cribriform pattern, mucincontaining cells, presence of psammoma bodies, and solid signetring cells. None of the histological parameters are specific for a particular genotype [14-19]. Fluorescence in situ hybridization using break-apart probes is useful to confirm ALK gene rearrangements; however, polymerase chain reaction and/or sequencing of RNA transcripts is required for delineating ALK fusion partners. ALK protein expression in a nuclear membranous or perinuclear cytoplasmic pattern gives a clue to the presence of underlying RANBP2 or RRBP1 fusion partners with ALK, which are the most common fusions in EIMS; however, these fusions can also be observed in IMTs and ALCLs [20]. On the other hand, other fusion partners including EML4, the most common fusion partner in ALK-rearranged NSCLC, have also been reported in EIMS associated with the more common cytoplasmic ALK staining pattern [26]. The various ALK gene rearrangement seen in NSCLC are EML4, TFG, KIF5B, KCL1, and multiple EML4 isoforms [27].

Differential diagnosis of poorly differentiated epithelioid to rhabdoid tumors
Thus, due to considerable genetic overlap with other ALKomas, EIMS is essentially a histopathological diagnosis, and definitive diagnosis can be difficult on a small biopsy, even with ancillary testing.
EIMS is an aggressive tumor with poor prognosis and local recurrence [1,3]. Review of previous reported pulmonary EIMS showed that surgical resection remained the mainstay of treatment when feasible (Table 1). These tumors do not respond to chemotherapy or radiotherapy [28,29]. Most ALK fusion proteins including those resulting from RANBP2-ALK fusions are sensitive to ALK tyrosine kinase inhibitor (TKI) inhibition in pre-clinical models [30], suggesting a role for targeted therapy. From the limited data available, EIMS does appear to respond to the ALK TKI crizotinib, as in our patient who experienced complete resolution of liver metastases. However, long-term responses or remissions are rare, with most EIMS patients progressing with metastatic disease within 3 to 6 months of crizotinib monotherapy [7-9], likely due to secondary resistance mechanisms [10]. A recent study demonstrated prolonged survival with a combination of ALK and CD30 targeted therapies [31]; however, CD30 expression appears to be uncommon in pulmonary EIMS (Table 1). EIMS is a very rare subtype of IMT that presents as a primary lung mass in young non-smokers, posing a considerable diagnostic challenge due to aggressive behavior with frequent metastases. There are limited data on treatment protocols; in the present case, targeted ALK inhibitor therapy combined with surgery achieved considerable disease control.
Notes
Ethics Statement
This study was approved by the Institutional Review Board with a waiver of informed consent (IRB No IEC-404/02.09.2016) and performed in accordance with the principles of the Declaration of Helsinki.
Availability of Data and Material
All data generated or analyzed during the study are included in this published article (and its supplementary information files).
Code Availability
Not applicable.
Author contributions
Conceptualization: PS. Data curation: PS. Formal analysis: PS. Supervision: DJ. Visualization: DJ. Writing—original draft: PS. Writing—review & editing: PS, AN, MKG, RR, SK, PSM, DJ. Approval of final manuscript: all authors.
Conflicts of Interest
D.J., a contributing editor of the Journal of Pathology and Translational Medicine, was not involved in the editorial evaluation or decision to publish this article. All remaining authors have declared no conflicts of interest.
Funding Statement
No funding to declare.