What’s new in dermatopathology 2023: WHO 5th edition updates
Article information

Abstract
The 5th edition WHO Classification of Skin Tumors (2022) has introduced changes to nomenclature and diagnostics. Important differences are discussed below. Changes in each category of skin tumor have been detailed, with particular emphasis on meaningful advances in our understanding of the molecular pathogenesis of the skin’s diverse tumor landscape.
KERATINOCYTIC/EPIDERMAL TUMORS
• Keratoacanthoma is kept separate from squamous cell carcinoma (SCC). Continued recognition that keratoacanthoma likely represents an SCC variant with self-resolving potential (Fig. 1).
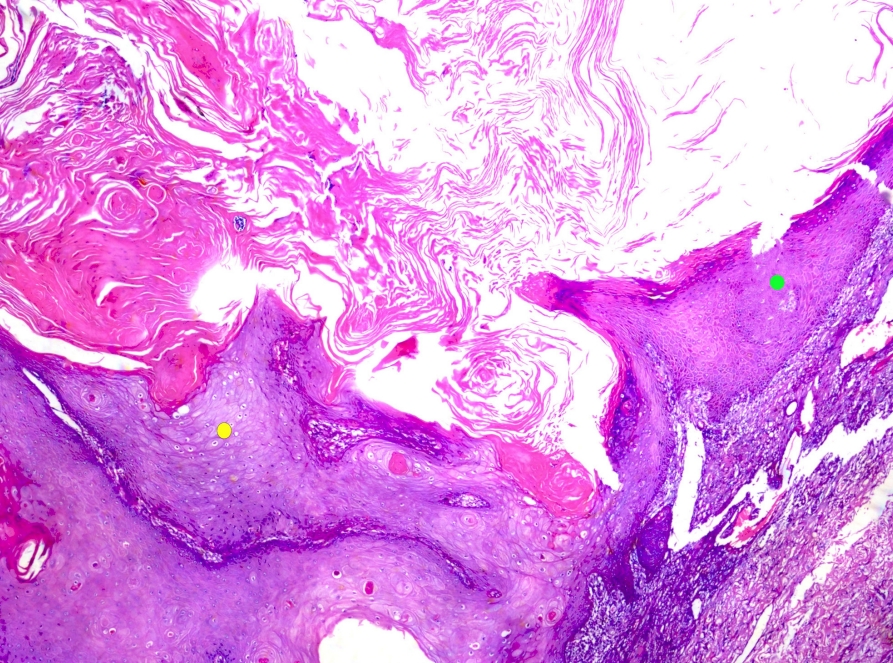
Keratoacanthoma. This crateriform squamous cell carcinoma variant possesses self-resolving potential. Thick glassy epithelium (yellow dot) may regress via thinner basophilic epithelium (green dot) as shown in the image.
• Adenosquamous carcinoma has been removed as an SCC subtype. It is thought to represent squamoid eccrine ductal carcinoma; discussed with adnexal neoplasms.
• Merkel cell carcinoma (Fig. 2) designated a neuroendocrine carcinoma of skin. In non-Merkel cell polyomavirus (MCPyV)-associated cases, TP53 and RB1 mutations seen in other neuroendocrine carcinomas are identified. Origin cell remains unclear.
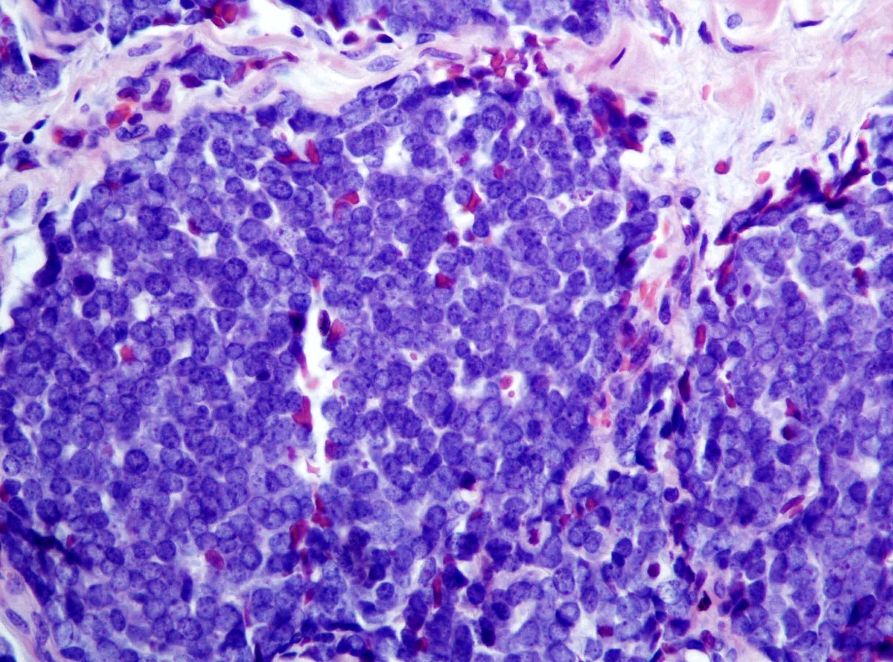
Merkel cell carcinoma (primary cutaneous neuroendocrine carcinoma of skin). The tumor demonstrates round blue cells with a characteristic finely dispersed “salt and pepper” chromatin pattern. Both Merkel cell polyomavirus associated and non-associated cases exist.
MELANOCYTIC NEOPLASMS
• Advanced understanding of the molecular pathways in melanocytic lesions have led to definition changes. Restrained proliferation of activated oncogenes by tumor suppressor genes leads to nevi, while additional mutations and escape from tumor suppressor gene control result in intermediate/malignant neoplasms [1].
• Nevi: clonal neoplasms with a single mutation, no other pathogenic changes, bland cytologic appearance and benign behavior.
• Melanocytomas: intermediate neoplasms between nevi and melanomas harboring > 1 driver mutation. Display atypical histopathologic features and potential for local recurrence. Second mutations affect specific pathways resulting in reproducible clinical and microscopic features.
• Some melanocytomas (previously designated “nevi”) have been renamed to reflect respective specific pathway aberrations and their intermediate status.
- Wnt-activated deep penetrating/plexiform melanocytoma (Fig. 3)
Wnt-activated deep penetrating/plexiform melanocytoma. This tumor lacks maturation with descent, displays nested/plexiform melanocytes with visible nucleoli, some nuclear pleomorphism, nuclear pseudoinclusions, and melanophages. Expresses nuclear β-catenin and LEF1.
- Pigmented epithelioid melanocytoma (PEM, also known as PRKAR1Ainactivated melanocytoma)
- BAP1-inactivated melanocytoma
- Spitz melanocytoma (previously atypical Spitz tumor)
- Microphthalmia-associated transcription factor (MITF) pathway-activated melanocytic tumors, see below
• Immunohistochemistry can aid in diagnosis:
- Loss of expression of PrkAr1a in some PEM
- Diffuse nuclear β-catenin, nuclear LEF1 in Wnt-activated deep penetrating/plexiform melanocytoma [2].
- Loss of nuclear BAP1 expression in BAP1-inactivated melanocytoma
• MITF pathway-activated melanocytic tumors are a newly introduced set of melanocytomas with cytoplasmic clearing and fusion genes resulting in overactive MITF functioning [3,4]. Main differentials include clear cell sarcoma, PEComa, melanoma and carcinomas. Two variants are described
- Clear cell tumor with melanocytic differentiation and ACTIN::MITF Translocation (CCTMAM)
° Cutaneous nodule
° Dermal based +/- subcutis. Marked cytoplasmic clearing. Low/high-grade nuclear features but mitoses are inconspicuous and the lesion lacks ulceration or perineural invasion. MART-1, HMB45, S100, MITF positive. Pankeratin negative.
- Clear cell tumor with melanocytic differentiation and MITF::CREM translocation (CCTMMC)
° Cutaneous nodule
° Dermal based +/- subcutis
° Marked cytoplasmic clearing (high-grade areas may lack clear cell change). Perineural invasion and increased mitotic rate described. No ulceration or vascular invasion reported.
° Diffuse MART1, S100, SOX10 and MITF. Patchy HMB45. Pankeratin negative.
• Language endorsements for lesions lacking clear diagnostic criteria
- Superficial atypical melanocytic proliferation of uncertain significance (SAMPUS) and intraepidermal atypical melanocytic proliferation of uncertain significance (IAMPUS) for lesions falling short of radial growth phase or in-situ melanoma, respectively. These designations imply virtually no risk of distant spread.
- Melanocytic tumor of uncertain malignant potential (MELTUMP) for neoplasms where vertical growth phase of melanoma is the main alternative and thus the uncertainty lies in potential metastatic risk.
ADNEXAL TUMORS
• Updates in molecular pathology of adnexal tumors
- Most poromas and some porocarcinomas harbor gene fusions YAP1::MAML2 or YAP1::NUTM1 [5]. Immunohistochemistry with NUT identifies those with NUTM1 rearrangements
- Some hidradenomas exhibit CRTC1::MAML2 fusion gene.
- ALPK1 mutations activating NF-κB pathway in some spiradenomas and spiradenocarcinomas (mutually exclusive from CYLD mutations).
- ETV6::NTRK3 translocation and NFIX::PKNI fusion in cutaneous secretory carcinoma.
• Cutaneous NUT carcinoma newly introduced (provisional)
- Rarely described BRD3::NUTM1 or NSD3::NUTM1 rearranged tumors [6].
- Dermal, infiltrating neoplasm arranged in islands, cords and/or nests.
- Glandular and squamoid differentiation with abrupt keratinization.
- Vesicular nuclei, prominent nucleoli.
- Positive NUT immunohistochemistry; CEA/EMA highlight ductules.
- Metastatic potential
• Cribriform carcinoma is renamed as cribriform tumor; definite malignant potential is unclear.
TUMORS OF THE NAIL UNIT
• Newly introduced section encompassing:
- Onychomatricoma (Fig. 4)
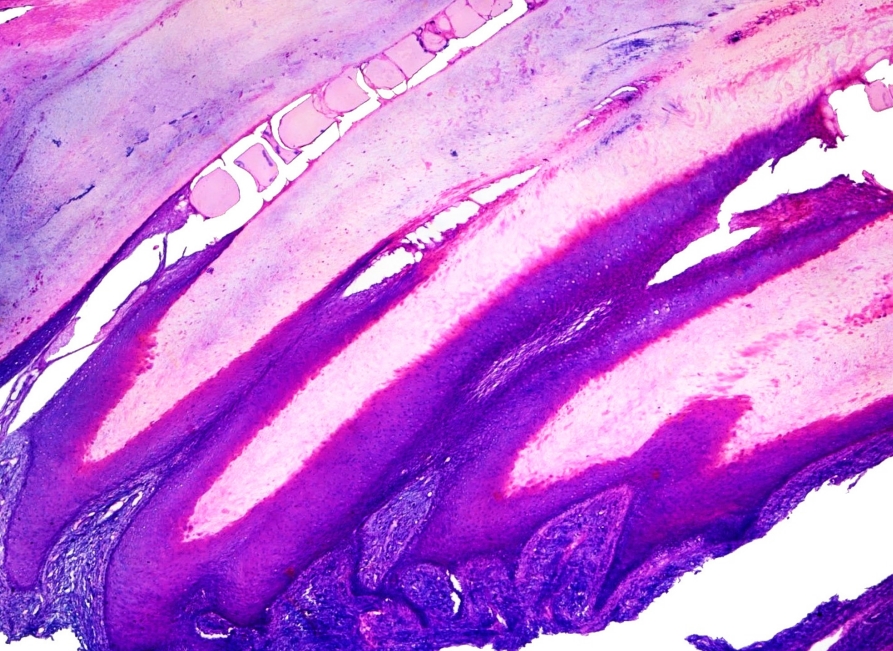
Onychomatricoma. Note typical papillomatous projections of matrical-type epithelium with deep V-shaped invaginations. Subjacent spindle cell proliferation expresses CD34.
- Onychopapilloma
- Ungual fibrokeratoma
- Onychocytic matricoma
- Subungual keratoacanthoma
TUMORS OF HEMATOPOIETIC AND LYMPHOID ORIGIN
• Dendritic cell neoplasms
- Introduction of mature plasmacytoid dendritic cell proliferation (MPDCP) associated with myeloid neoplasm.
- MPDCP is a proliferation of plasmacytoid dendritic cells with low-grade cytology occurring in patients with known myeloid neoplasms (most commonly chronic myelomonocytic leukemia and acute myeloid leukemia).
• Histiocytic neoplasms
- ALK-positive histiocytosis: A histiocytic neoplasm that may histologically resemble juvenile xanthogranuloma and is characterized by ALK gene rearrangement and positive ALK immunohistochemistry.
- Indeterminate dendritic cell tumor replaces indeterminate dendritic cell “histiocytosis”. The definitive cell of origin remains unclear.
- BRAF V600E mutations in Langerhans cell histiocytosis increase risk of relapse, severe clinical manifestations and treatment failure [7]. Patients may benefit from targeted BRAF inhibitor therapy.
• T-cell and NK-cell lymphoproliferative disorders and neoplasms
- Primary cutaneous T-cell lymphomas (PCTCL) are all listed as individual entities including those previously labeled “rare subtypes”, i.e., subcutaneous panniculitis-like T-cell lymphoma, extranodal NK/T-cell lymphoma, primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma, primary cutaneous γ/δ T-cell lymphoma, primary cutaneous CD4-positive small or medium T-cell lymphoproliferative disorder, and primary cutaneous acral CD8- positive lymphoproliferative disorder.
- Cutaneous CD8-positive acral T-cell lymphoma reclassified as a lymphoproliferative disorder. This entity is indolent with excellent long-term outcomes.
- “Primary cutaneous peripheral T-cell lymphoma, not otherwise specified (NOS)” should be used for cases that cannot be classified as a known PCTCL subtype.
- Newly introduced section on inborn error of immunity-associated lymphoproliferative disorders, which are CD8+ T-cell rich dermal infiltrates +/- granulomatous inflammation associated with inborn immunodeficiencies. May present as nodules, papules or ulceronecrotic lesions. EBV in-situ hybridization must be negative.
SOFT TISSUE TUMORS
• Introduction of four new entities:
- CRTC1::TRIM11 cutaneous tumor: dermal-based neoplasm characterized by well-circumscribed proliferation of spindled/epithelioid cells with pale cytoplasm. Arranged in nests/fascicles and may or may not have a vaguely palisaded appearance. Diffuse SOX10 expression; variable positivity for S100 and other melanocytic markers. CRTC1::TRIM11 fusion. Most commonly seen on the extremities. Generally indolent but may recur locally or metastasize [8,9].
- Superficial CD34-positive fibroblastic tumor: indolent neoplasm occurring in the skin and subcutis with a predilection for the extremities. Characterized by a mixture of spindled and epithelioid cells with moderate pleomorphism, prominent nucleoli, nuclear pseudoinclusions and eosinophilic cytoplasm (granular/glassy). Admixed mixed inflammatory cells are characteristic. Diffuse CD34, PRDM10, and CADM3 expression. AE1/AE3 positivity >50%. PRDM10 rearrangement in >50% of cases [10]. Excision is curative in most cases.
- EWSR1::SMAD3 rearranged fibroblastic tumor: well-demarcated superficial tumor exhibiting intersecting fascicles of bland fibroblastic spindle cells peripherally and relatively acellular, hyalinized collagen centrally (may lack clear zones). Diffuse nuclear ERG positivity; negative for SMA, EMA, SOX10, CD34, S100. EWSR1::SMAD3 fusion present. Benign tumor; may have local recurrence.
- NTRK-rearranged spindle cell neoplasm: group of spindle cell lesions with frequent NTRK rearrangements, most commonly seen in children. Involves the dermis and subcutis with a spectrum of appearances, including bland spindle cells in fibrous septa entrapping mature fat to highly cellular lesions with sheets/fascicles of spindle cells. Foci of high-grade atypia is possible. Variable CD34, SMA, S100 positivity. Diffuse panTRK positivity if NTRK fusion present.
• Atypical intradermal smooth muscle neoplasm remains the preferred terminology for smooth muscle tumors with cytologic atypia limited to the dermis. Lesions have limited metastatic potential and an excellent prognosis once completely excised.
• Epithelioid fibrous histiocytoma is classified as being of uncertain differentiation rather than a dermatofibroma subtype.
GENETIC TUMOR SYNDROMES ASSOCIATED WITH SKIN MALIGNANCIES
Newly introduced section detailing tumor syndromes with cutaneous neoplasms including:
• Familial melanoma
• BAP1 tumor predisposition syndrome
• Xeroderma pigmentosum
• Nevoid basal cell carcinoma syndrome (Gorlin syndrome)
• Carney complex
• Muir-Torre syndrome
• Brooke-Spiegler and related syndromes
Meet the Authors
Dr. Jonathan Ho has been an author for PathologyOutlines.com since 2021 and the Deputy Editor for Dermatopathology since January 2023. He is currently a Lecturer at The University of the West Indies, Mona Campus, Jamaica where he practices dermatopathology and dermatology and is the CoDirector of the dermatology residency program.
Dr. Chico Collie has been a resident author for PathologyOutlines.com since 2021. He is the Chief Resident in Pathology at the University of the West Indies, Mona Campus, Jamaica. He is passionate about surgical pathology, with a subspecialty interest in dermatopathology.