Lymphomatoid papulosis: a practical review for pathologists
Article information

Abstract
Lymphomatoid papulosis (LyP) is a primary cutaneous CD30+ lymphoproliferative disorder characterized by a chronic and self-healing recurrent cluster of erythematous papules or nodules on the skin of the trunk and/or extremities. The disease has an indolent clinical course with spontaneous regression or waxing and waning clinical evolution. The histopathologic spectrum of LyP is vast and may show few to numerous atypical cells immersed in a mild to intense inflammatory background. The backbone for the diagnosis is the positivity for CD30, which is one of the criteria to define this group of lymphoproliferative disorders. The association of these different histological and immunophenotypical findings is used to subclassify this disease in different subtypes from A to E, associated with DUSP22/IRF4 rearrangement, and other rare forms. Although this differentiation is important to raise awareness of different differential diagnosis, it does not impact the prognosis or change the treatment, which is usually centered in symptom relief and faster regression. In this review, we aim to summarize the most updated information of the clinical, histopathological, and molecular characteristics of LyP and provide a practical assessment for the diagnostic features that could help with the main differential diagnosis.
INTRODUCTION
Lymphomatoid papulosis (LyP) is a chronic and self-healing lymphoproliferative disorder of the skin and is mainly characterized by recurrent and self-healing erythematous papules or nodules on the trunk or extremities [1-8]. This entity is currently characterized under the group of CD30+ lymphoproliferative disorders by both the 5th edition of the World Health Organization of Hematolymphoid Tumors and the International Consensus Classification [2,9]. It was first described in 1968 by Dr. W. Macaulay, who reported a 41-year-old patient with waxing and waning papules for 3 years, for which the biopsy showed an intense infiltrate of atypical cells, suggesting a lymphoma in the skin [10].
Primary cutaneous CD30+ lymphoproliferative disorders include primary cutaneous anaplastic large cell lymphoma (pc-ALCL) and LyP and account for 25% of all cutaneous T-cell lymphomas [5,11]. Although LyP is a relatively frequent condition among the primary cutaneous T-cell lymphomas, this is a very rare condition in the general population and has an incidence of 1.2–1.9 cases per 1 million people [12].
The incidence peak occurs in the fifth decade but can affect all age groups [1,3,13] and it is more common in men than women [1,13,14]. Pediatric cases have been reported and have a similar presentation to the adult ones [15]. Overall, it is also more common in white populations [5] and LyP type A is the most common subtype, followed by type D and type B [14], although LyP type B is more common in Hispanics [1].
ETIOPATHOGENESIS AND CLINICAL FEATURES
The etiopathogenesis of LyP is unknown, and no viral etiologic factors, such as Epstein-Barr virus, are associated with the development or progression of the disease. Some cases that show progression to anaplastic large cell lymphoma show resistance to CD30 ligand and alterations involving the TGF-β signaling pathway [5].
Most patients are previously asymptomatic and lack underlying disease, while a subset of cases arises in patients with concurrent or a history of mycosis fungoides (MF) [16]. The typical clinical presentation is characterized by clusters of red or purple papules and small nodules (<2.0 cm each) with different stages of development, spontaneous regression with crusts in a few weeks and an indolent clinical course [1,5,8,11,17,18]. Less commonly, single or multiple crops are observed, and the disease can persist from several weeks to years [1,5,18]. The degree of lesion severity is measured by the number of lesions during the clinical presentation, being defined as mild (<12 lesions), moderate (12–50 lesions), and severe (>50 lesions) [1]. The lesions are more common in the trunk and extremities but can affect any body part [1,3,19] (Fig. 1A). Oral and mucosal involvement is rare [5] and may be accompanied by ulceration and necrosis [20]. The development of secondary lymphomas may precede, occur concurrently with, or follow the diagnosis and is reported in 10% to 20% of the patients [21,22].
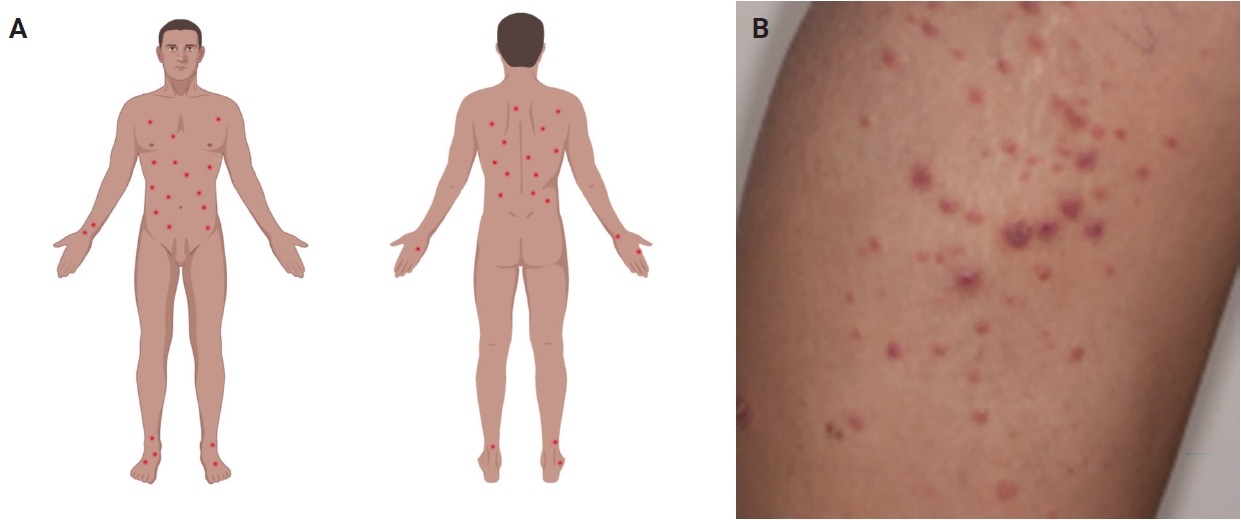
(A) The classic clinical presentation of lymphomatoid papulosis (LyP) is characterized by clustered erythematous papules mainly distributed on the trunk and extremities (image created with Biorender.com). (B) Clinical image displaying a cluster of macules and papules that are characteristic of LyP. Note that some papules show central crusting, consistent with involuting lesions.
The dermoscopic appearance of these lesions may coexist in different stages of progression and involution [17,23,24], including: (1) pinkish or light brown papules with homogeneous areas (Fig. 1B) and delicate dotted vascular pattern at the periphery; (2) papules with centripetal and tortuous irregular vessels; (3) crusts and papules with central scale and vascular pattern sparing the center in mature lesions; (4) papule with central necrotic ulceration denotes old lesion; (5) postinflammatory pigmentation in lesional areas; and (6) pruritus as the primary symptom in about half of patients [1]; among other more rare patterns.
A correct diagnosis requires an adequate clinical history of papular lesions with spontaneous regression associated with typical histological and immunohistochemical findings [1,3,5,7].
HISTOPATHOLOGICAL FINDINGS
Histopathological presentation in LyP is characterized by a subtle wedge-shaped distribution of the infiltrate, with a wide superficial base and a tapered tip extending into the deep dermis and less frequently into the subcutaneous tissue [25] (Fig. 2A). The wedge-shaped infiltrate may not be readily apparent in a punch biopsy and requires an excisional biopsy for better visualization. The most characteristic appearance is the presence of few to numerous large cells with a Hodgkin or Hodgkin/Reed-Sternberg–like cells mixed with a reactive background of small lymphocytes and less frequently eosinophils, plasma cells, and histiocytes [25] (Fig. 2B). Some cases display pseudoepitheliomatous hyperplasia that may raise the possibility of squamous cell carcinoma [26].
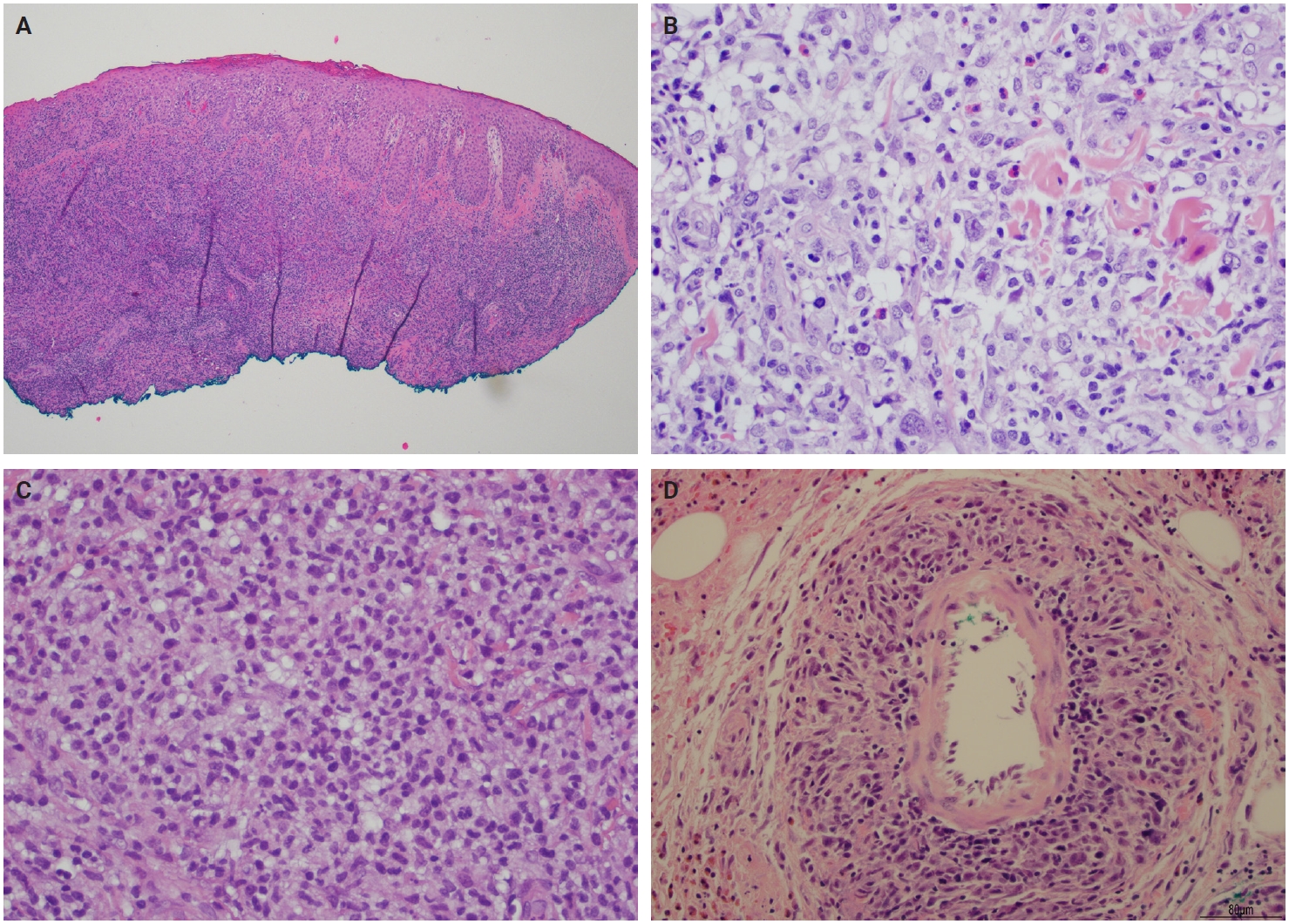
Lymphomatoid papulosis (Lyp). (A) Histological section of LyP shows a dense and diffuse dermal infiltrate with focal epidermotropism. (B) Histological section of LyP type A shows Hodgkin/Reed-Sternberg–like cells in a background of small lymphocytes, eosinophils, and histiocytes. (C) LyP type C shows a diffuse infiltrate of intermediate to large lymphocytes with moderately abundant cytoplasm, large vesicular nuclei, and distinct nucleoli. (D) LyP type E is characterized by angiotropism as shown in this medium-sized vessel extensively infiltrated by large abnormal lymphocytes.
The microscopic architecture and immunophenotype of large cells and their background are variable and have led to subclassification of LyP into different categories [1,5,6,13,14,27-33]. In LyP type A, neoplastic CD30+ cells are scattered and admixed with an extensive lymphoid infiltrate of neutrophils, eosinophils, and histiocytes mimicking classic Hodgkin lymphoma (Fig. 2B). In LyP type B, the presentation is characterized by epidermotropism and band-like distribution of small to medium atypical lymphocytes with cerebriform nuclei, usually CD30-negative, and mimicking MF, patch stage. Type C of LyP is composed of sheets of large neoplastic cells, with or without epidermotropism that display uniform positivity for CD30, mimicking a pc-ALCL; in this type, only few admixed inflammatory cells are noted. (Fig. 2C). In contrast, LyP type D presents with atypical small to medium-sized lymphocytes with distinct epidermotropism and these neoplastic cells express CD30 and CD8 and mimic pagetoid reticulosis. LyP type E shows an angiocentric and angiodestructive pleomorphic lymphoid infiltrate of small- to medium-sized lymphocytes admixed with scattered large cells uniformly positive for CD30 and the overall appearance is that of an aggressive lymphoma, such as extranodal T/natural killer (NK) cell lymphoma (Fig. 2D). LyP with DUSP22/IRF4 rearrangement (please note that it is not DUSP22::IRF4 since there is not a rearrangement between DUSP22 and IRF4; they are genomically too close for fluorescence in situ hybridization to separate them, so if positive, the rearrangement can be for DUSP22 or IRF4) displays a dense diffuse lymphoid infiltrate (Fig. 3A). A biphasic growth pattern with pagetoid reticulosis-like epidermotropism of small to medium size cerebriform lymphocytes (Fig. 3B) that express dim or lack CD30, while the second component is dermal or periadnexal (Fig. 3C) and the atypical lymphocytes express bright CD30+ (Fig. 3D) [25].
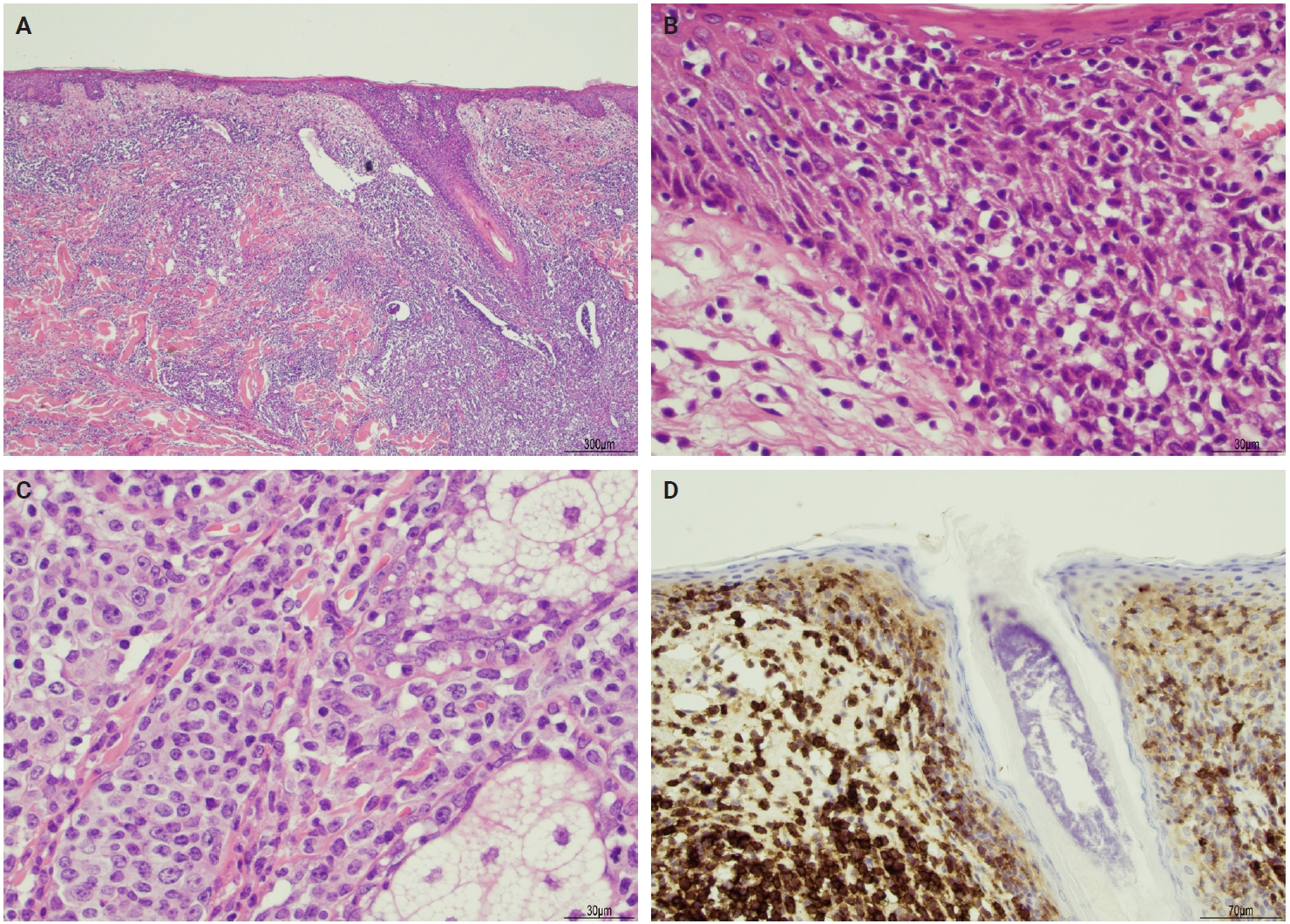
Lymphomatoid papulosis (LyP) with DUSP22/IRF4. (A) Histological section shows dense lymphoid infiltrate in the dermis with epidermotropic infiltrate. (B) High magnification shows a predominance of small epidermotropic lymphocytes. (C) High magnification shows a predominance of large cells in the dermis. (D) Immunohistochemistry for CD30 highlights the biphasic pattern of this LyP variant displaying faint reactivity in epidermotropic lymphocytes while there is strong CD30 reactivity in the dermal lymphocytes. Courtesy of Dr. Sofia A. Garces.
Rare forms of LyP presentation include folliculotropic, syringotropic, granulomatous, and gamma-delta variant. In all cases, large atypical CD30+ cells are identified. In the folliculotropic pattern, there is a perifollicular infiltrate of atypical lymphocytes, cystic dilatation sometimes with rupture of the hair follicle, hyperplasia of the follicular epithelium, intrafollicular pustules (neutrophil collections), and follicular mucinosis [34]. The syringotropic pattern is characterized by eccrine units with periglandular infiltrate [14], while the granulomatous LyP shows mononuclear infiltrate with perivascular, eccrinotropic, and neurotropic distribution associated with non-caseating granulomas [14,35]. The gamma-delta variant may mimic aggressive lymphomas such as primary cutaneous gamma-delta T-cell lymphoma or MF. Most of the cases show cytotoxic immunophenotype; however, the clinical presentation is more akin to LyP [14,36]. Different histologic subtypes may coexist simultaneously or be present in the same patient throughout the clinical evolution of the lesions and even be associated with other CD30+ lymphoproliferative disorders such as pc-ALCL [37].
IMMUNOHISTOCHEMICAL AND ANCILLARY STUDIES
The neoplastic cells of LyP usually express CD3, CD4, CD25, CD30, CD45RO, human leukocyte antigen–DR isotype (HLA-DR), T-cell intracellular antigen-1 (TIA1), and granzyme B consistent with an activated T helper immunophenotype. A variable loss of T-cell antigens may be seen. In effect, these cells are characterized by the positivity for CD3 and CD4 as T helper cells. CD45RO defines them as memory/activated T cells while CD25, CD30, and HLA-DR define them as activated lymphocytes. Lastly, TIA1 and granzyme B indicate their cytotoxic phenotype [5,6,24,27-30]. Other T-cell antigens, such as CD2, CD5, and CD7, have a variable expression including complete negativity, depending on the subtype of LyP. Although CD8 is negative in LyP types A, B, and C, it is usually positive in LyP type D and has a variable positivity in type E and DUSP22/IRF4 rearrangement cases. The biomarker scenario of LyP is mainly characterized by the CD30 expression in the neoplastic cells, helping not only in the diagnostic definition of the entity, but also with the possibilities of new therapeutic options. Table 1 summarizes the main immunophenotypical features of different subtypes of LyP.
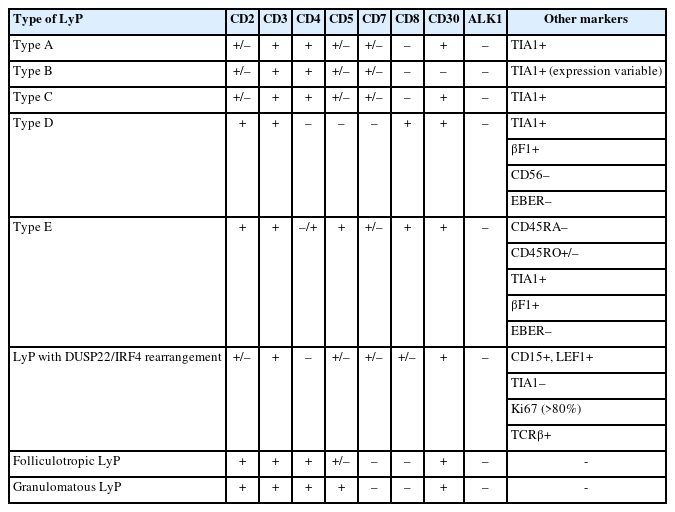
Immunohistochemical summary of LyP types
Monoclonal rearrangements of the T-cell receptor gamma (TRG) or beta (TRB) chains are usually detected [5] and may overlap with other coexisting lymphomas, such as MF and pc-ALCL [38]. Recurrent mutations in epigenetic modifying genes (SETD2, KMT2A, KMT2D, and CREBBP) are the most frequently seen, but no specific mutations are observed, since a similar mutational profile is seen in pc-ALCL [39]. Recurrent NPM1::TYK2 gene fusions in LyP induce activation of the STAT signaling pathway [40,41]. More recently, cases with characteristic DUSP22/IRF4 rearrangement/6p25.3 translocation cases have been described and show characteristic features, such as common presentation in older patients, localized lesions with clinical presentation suggesting benign inflammatory dermatoses or epithelial tumors, and histological presentation with a bimodal pattern. The epidermotropic lymphocytes are small and are negative or express weak CD30, while the dermal cells are large, express strong CD30 and lymphocyte enhancer binding-factor 1, and are negative for TIA1 [6].
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of LyP is broad due to the great overlap in the clinical, histological, and immunophenotypical presentations with malignant skin diseases as well as benign dermatoses. The main differential diagnoses according to the specific type of LyP are summarized in Tables 2 and 3.
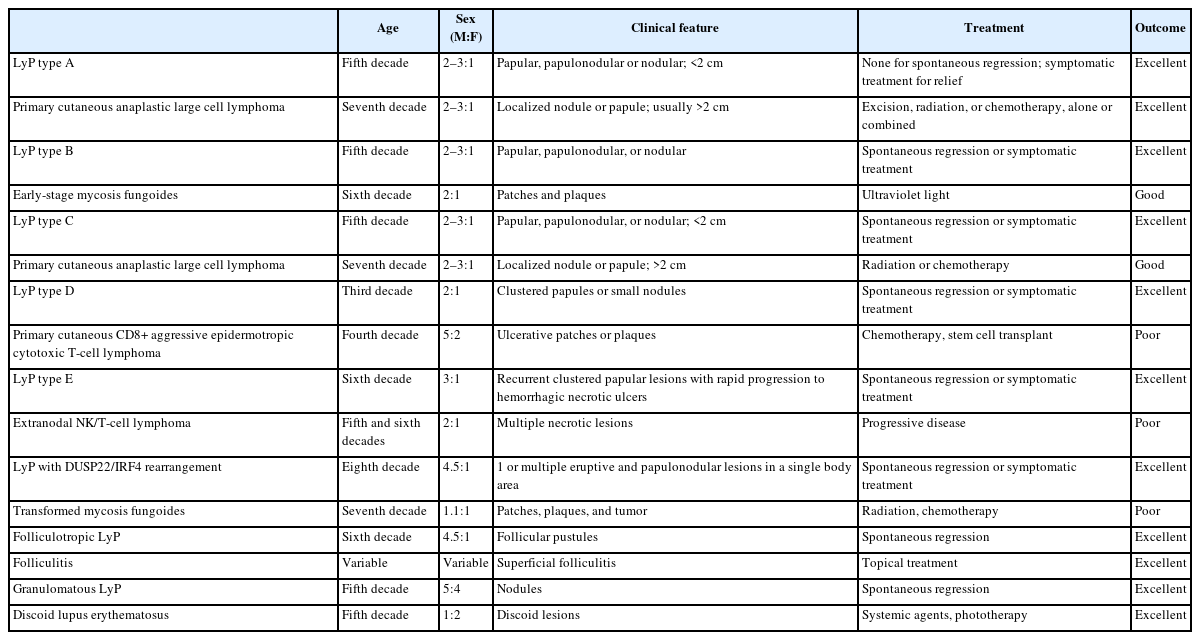
Main clinical features of differential diagnoses of different subtypes of LyP
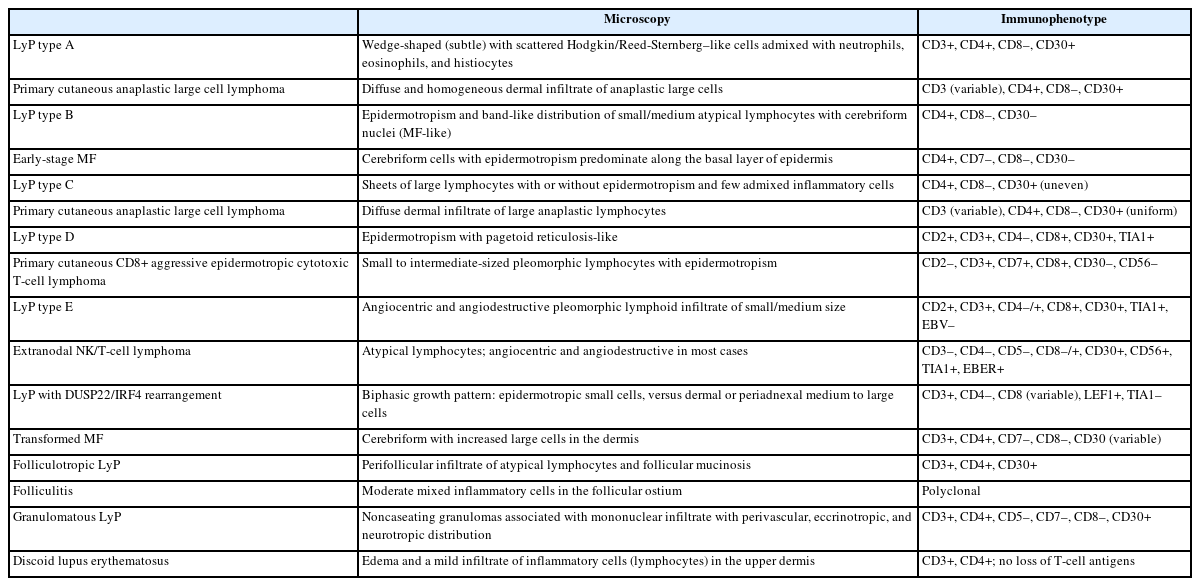
Main pathologic and immunophenotypic features of differential diagnoses of different subtypes of LyP
Primary cutaneous anaplastic large cell lymphoma (pc-ALCL)
Pc-ALCL is the main differential diagnosis of LyP type A and type C [27,42]. Both LyP and pc-ALCL are more prevalent in young adults. In LyP, the clinical presentation is characterized by multiple papules, papulonodular or nodular lesions <2 cm, while in pc-ALCL localized or single nodules or papules more than 2 cm are more common. The histological presentation in pc-ALCL is characterized by diffuse and homogeneous dermal infiltration of anaplastic cells without epidermotropism, while in LyP could vary from polymorphous and wedge-shaped in LyP type A and sheets of large lymphocytes with or without epidermotropism with few admixed inflammatory cells in type C. Cases of LyP type C could be morphologically identical to pc-ALCL, and because of this, the clinical correlation is fundamental for the final diagnosis. Both entities have a T-helper immunophenotype, characterized by CD3+ and CD4+, negativity for CD8, and diffuse positivity for CD30. Cases of pc-ALCL can also harbor DUSP22/IRF4 gene rearrangements, while cases of LyP usually express multiple myeloma oncogene 1/IRF4 and are negative for anaplastic lymphoma kinase-1, being additional markers that could help in the differential diagnosis [43,44]. The outcome is excellent in both entities [27,42].
Early-stage MF
Cases of LyP type B could have some overlapping histological and immunophenotypical presentation with early-stage MF. Both neoplasms present in adults and have a slight predominance in males. The clinical presentation in LyP is characterized by papular, papulonodular, or nodular lesions, while early-stage MF presents with patches and plaques. Histologically, an epidermotropic infiltrate of small/medium cerebriform cells is observed in both entities and might be indistinguishable. The immunophenotype is also very similar in both, being CD3+, CD4+, CD8–, and CD30–. The clinical correlation is fundamental to precisely determine the diagnosis [27,45].
Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma
This is a rare and clinically aggressive primary cutaneous lymphoma with a poor prognosis, which may have overlapping features with LyP type D. Both affect young adult men; however, the clinical presentation of primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma (PC CD8-AECTCL) is more dramatic and the lesions are usually ulcerative patches or plaques with rapid progression and necrosis. The main immunophenotypic difference is centered on the CD30 expression, which is positive in LyP and negative in PC CD8-AECTCL; however, the cytotoxic immunophenotype (CD3+CD4–CD8+) is common in both [30,46].
Extranodal NK/T-cell lymphoma involving skin
LyP type E is the main differential diagnosis of extranodal NK/T-cell lymphoma (ENKTCL). Both affect more men in the fifth to sixth decade of life. While LyP type E presents with recurrent clustered papular lesions with rapid progression to hemorrhagic necrotic ulcers, ENKTCL presents with multiple necrotic lesions. The histological presentation can be identical in both, characterized by angiocentric and angiodestructive pleomorphic lymphoid infiltrate of small to medium size and significant inflammatory background. The main immunophenotypical difference is centered on the association with Epstein-Barr virus, which is present in ENKTCL and negative in LyP type E. ENKTCL is also very aggressive and has a bad prognosis [6,27,47].
Large cell transformation of MF
Both LyP with DUSP22/IRF4 rearrangement and large cell transformation of MF affect elderly men. LyP with DUSP22/IRF4 presents with one or multiple eruptive and papulonodular lesions in a single body area, while patients with large cell transformation of MF refer to a history of evolution of patches, plaques, and tumors. This LyP subtype usually displays a biphasic growth pattern, where the epidermotropic component is that of small cells and the dermal/periadnexal component is composed of medium to large cells. Large cell transformation of MF has sheets of large cells, occasionally admixed with cerebriform lymphocytes. The immunophenotype is similar in both entities and CD30 tends to be more uniform in cases of LyP, but a diffuse and strong positivity cannot exclude large cell transformation of MF. The clinical correlation is fundamental to rendering the final diagnosis [6,48,49].
Additional differential diagnosis
Due to the broad histological and clinical manifestation of LyP, other entities that could also be considered as differential diagnosis, but will not be contemplated in this review, are (1) pityriasis lichenoides/pityriasis lichenoides et varioliformis acuta; (2) arthropod bite reaction; (3) reactive CD30+ cutaneous infiltrates; (4) systemic ALCL involving skin; and (5) classic Hodgkin lymphoma involving skin.
TREATMENT AND PROGNOSTIC FACTORS
Most patients do not require specific treatment [1,6,7,18], and clinical observation only is enough. Most of the treatments are aimed at symptom relief and faster regression. There are several treatment modalities, such as (1) low-dose methotrexate [11,18,31]; (2) psoralen ultraviolet A [1,7,11,18], which is related to longer disease-free survival: 10–36 months [18]; (3) topical corticosteroids [1,7,18]; (4) brentuximab vedotin, an active and well tolerated option and has shown a very high response rate in some reports [1,4]; (5) bexarotene [1]; (6) topical ruxolitinib [41]; and (7) topical alkylating agents (e.g., chlormethine gel) [50]. Treatment discontinuation often led to relapses and therapies often suppress lesions without altering the long-term disease course. Since the clinical course and the low necessity of treatment, few innovative therapeutic options are studied in clinical trials (https://clinicaltrials.gov/search?cond=Lymphomatoid%20Papulosis&viewType=Card).
The prognosis is excellent [5,7,11,13], and only around 5% of the patients die of disease over a median period of 4 years, particularly patients with established underlying cutaneous lymphoma [1]. Although it is important to differentiate different subtypes of LyP for a proper differential diagnosis, the histologic types do not have prognostic significance [27].
Patients with a diagnosis of LyP are recommended clinical follow-up, since there is a risk of developing a second lymphoid malignancy (e.g., MF and Hodgkin lymphoma) [4,5,7,11,13,18]. Patients with monoclonal TRB or TRG gene rearrangements or a mixed type of LyP have a higher risk of progressing to lymphoma [13], and an increased risk of subsequent lymphoma has been reported in men and in patients with LyP types B and C, while LyP type A and D are less frequently associated with lymphoma development [1]. Cases of LyP type A are the most prevalent subtype associated with early relapses [18] and elevated serum lactate dehydrogenase is associated with a high risk of associated lymphoproliferative disorders in all cases of LyP [8].
Notes
Ethics Statement
Not applicable.
Availability of Data and Material
Data sharing not applicable to this article as no datasets were generated or analyzed during the study.
Code Availability
Not applicable.
Author Contributions
Writing—original draft: MLMP, CATC, RNM. Writing—review & editing: MLMP, CATC, RNM. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.