 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 60(4); 2026 > Article
-
Review Article
Lymphomatoid papulosis: a practical review for pathologists -
Mario L. Marques-Piubelli1
 , Carlos A. Torres-Cabala2, Roberto N. Miranda3
, Carlos A. Torres-Cabala2, Roberto N. Miranda3 -
Journal of Pathology and Translational Medicine 2026;60(4):388-397.
DOI: https://doi.org/10.4132/jptm.2026.06.09
Published online: July 15, 2026
1The Healthcare Business of Merck KGaA, Darmstadt, Germany
2Department of Pathology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
3Department of Hematopathology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
- Corresponding Author: Roberto N. Miranda, MD Department of Hematopathology, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030, USA Tel: +1-713-745-2535, Fax: +1-713-794-1800 E-mail: Roberto.miranda@mdanderson.org
This article has been published jointly, with consent, in both Journal of Pathology and Translational Medicine and PathologyOutlines.com.
© The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 128 Views
- 9 Download
Abstract
- Lymphomatoid papulosis (LyP) is a primary cutaneous CD30+ lymphoproliferative disorder characterized by a chronic and self-healing recurrent cluster of erythematous papules or nodules on the skin of the trunk and/or extremities. The disease has an indolent clinical course with spontaneous regression or waxing and waning clinical evolution. The histopathologic spectrum of LyP is vast and may show few to numerous atypical cells immersed in a mild to intense inflammatory background. The backbone for the diagnosis is the positivity for CD30, which is one of the criteria to define this group of lymphoproliferative disorders. The association of these different histological and immunophenotypical findings is used to subclassify this disease in different subtypes from A to E, associated with DUSP22/IRF4 rearrangement, and other rare forms. Although this differentiation is important to raise awareness of different differential diagnosis, it does not impact the prognosis or change the treatment, which is usually centered in symptom relief and faster regression. In this review, we aim to summarize the most updated information of the clinical, histopathological, and molecular characteristics of LyP and provide a practical assessment for the diagnostic features that could help with the main differential diagnosis.
- Lymphomatoid papulosis (LyP) is a chronic and self-healing lymphoproliferative disorder of the skin and is mainly characterized by recurrent and self-healing erythematous papules or nodules on the trunk or extremities [1-8]. This entity is currently characterized under the group of CD30+ lymphoproliferative disorders by both the 5th edition of the World Health Organization of Hematolymphoid Tumors and the International Consensus Classification [2,9]. It was first described in 1968 by Dr. W. Macaulay, who reported a 41-year-old patient with waxing and waning papules for 3 years, for which the biopsy showed an intense infiltrate of atypical cells, suggesting a lymphoma in the skin [10].
- Primary cutaneous CD30+ lymphoproliferative disorders include primary cutaneous anaplastic large cell lymphoma (pc-ALCL) and LyP and account for 25% of all cutaneous T-cell lymphomas [5,11]. Although LyP is a relatively frequent condition among the primary cutaneous T-cell lymphomas, this is a very rare condition in the general population and has an incidence of 1.2–1.9 cases per 1 million people [12].
- The incidence peak occurs in the fifth decade but can affect all age groups [1,3,13] and it is more common in men than women [1,13,14]. Pediatric cases have been reported and have a similar presentation to the adult ones [15]. Overall, it is also more common in white populations [5] and LyP type A is the most common subtype, followed by type D and type B [14], although LyP type B is more common in Hispanics [1].
INTRODUCTION
- The etiopathogenesis of LyP is unknown, and no viral etiologic factors, such as Epstein-Barr virus, are associated with the development or progression of the disease. Some cases that show progression to anaplastic large cell lymphoma show resistance to CD30 ligand and alterations involving the TGF-β signaling pathway [5].
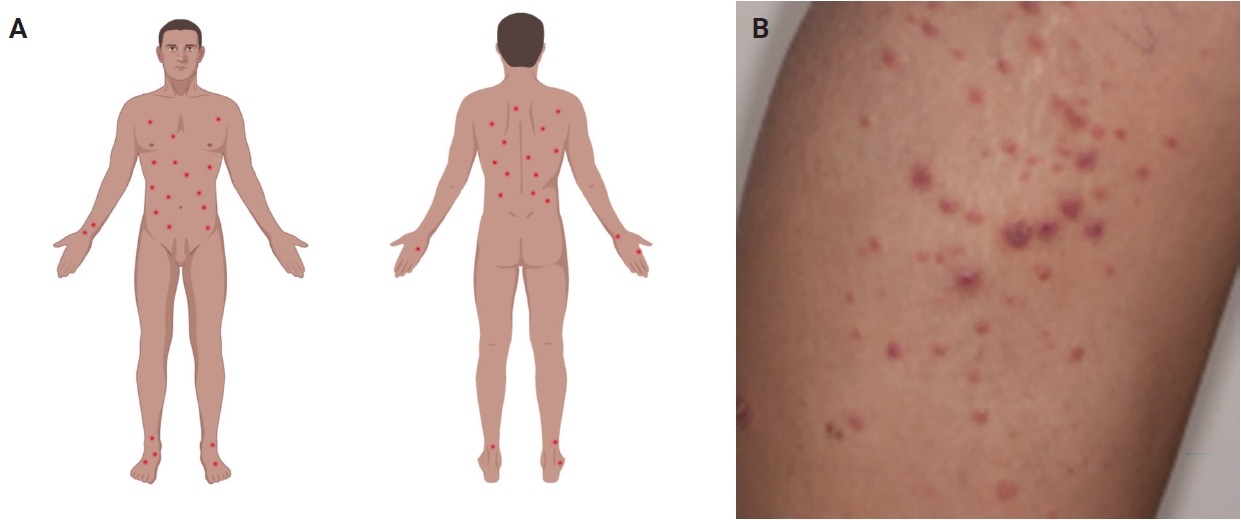
- Most patients are previously asymptomatic and lack underlying disease, while a subset of cases arises in patients with concurrent or a history of mycosis fungoides (MF) [16]. The typical clinical presentation is characterized by clusters of red or purple papules and small nodules (<2.0 cm each) with different stages of development, spontaneous regression with crusts in a few weeks and an indolent clinical course [1,5,8,11,17,18]. Less commonly, single or multiple crops are observed, and the disease can persist from several weeks to years [1,5,18]. The degree of lesion severity is measured by the number of lesions during the clinical presentation, being defined as mild (<12 lesions), moderate (12–50 lesions), and severe (>50 lesions) [1]. The lesions are more common in the trunk and extremities but can affect any body part [1,3,19] (Fig. 1A). Oral and mucosal involvement is rare [5] and may be accompanied by ulceration and necrosis [20]. The development of secondary lymphomas may precede, occur concurrently with, or follow the diagnosis and is reported in 10% to 20% of the patients [21,22].
- The dermoscopic appearance of these lesions may coexist in different stages of progression and involution [17,23,24], including: (1) pinkish or light brown papules with homogeneous areas (Fig. 1B) and delicate dotted vascular pattern at the periphery; (2) papules with centripetal and tortuous irregular vessels; (3) crusts and papules with central scale and vascular pattern sparing the center in mature lesions; (4) papule with central necrotic ulceration denotes old lesion; (5) postinflammatory pigmentation in lesional areas; and (6) pruritus as the primary symptom in about half of patients [1]; among other more rare patterns.
- A correct diagnosis requires an adequate clinical history of papular lesions with spontaneous regression associated with typical histological and immunohistochemical findings [1,3,5,7].
ETIOPATHOGENESIS AND CLINICAL FEATURES
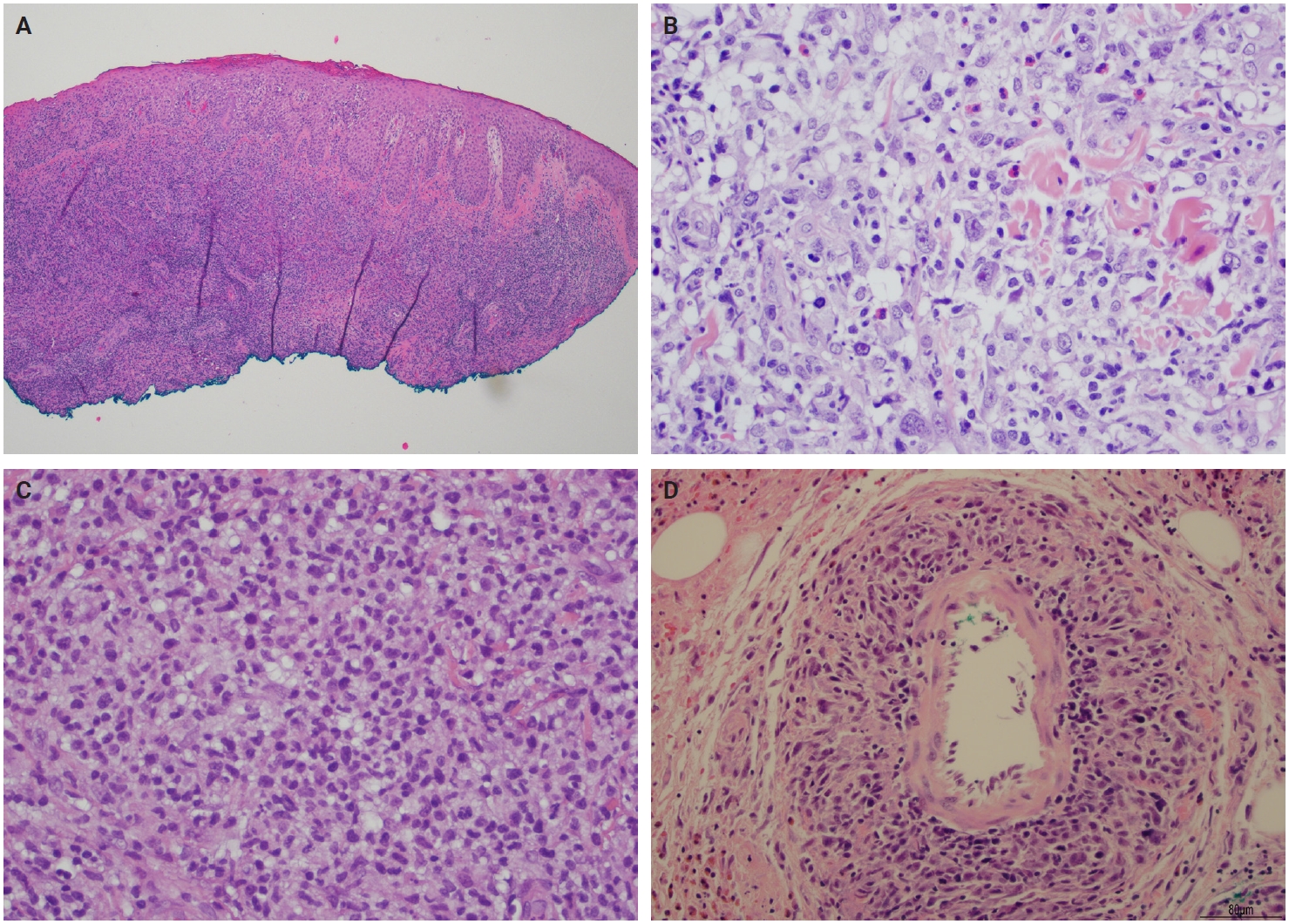
- Histopathological presentation in LyP is characterized by a subtle wedge-shaped distribution of the infiltrate, with a wide superficial base and a tapered tip extending into the deep dermis and less frequently into the subcutaneous tissue [25] (Fig. 2A). The wedge-shaped infiltrate may not be readily apparent in a punch biopsy and requires an excisional biopsy for better visualization. The most characteristic appearance is the presence of few to numerous large cells with a Hodgkin or Hodgkin/Reed-Sternberg–like cells mixed with a reactive background of small lymphocytes and less frequently eosinophils, plasma cells, and histiocytes [25] (Fig. 2B). Some cases display pseudoepitheliomatous hyperplasia that may raise the possibility of squamous cell carcinoma [26].
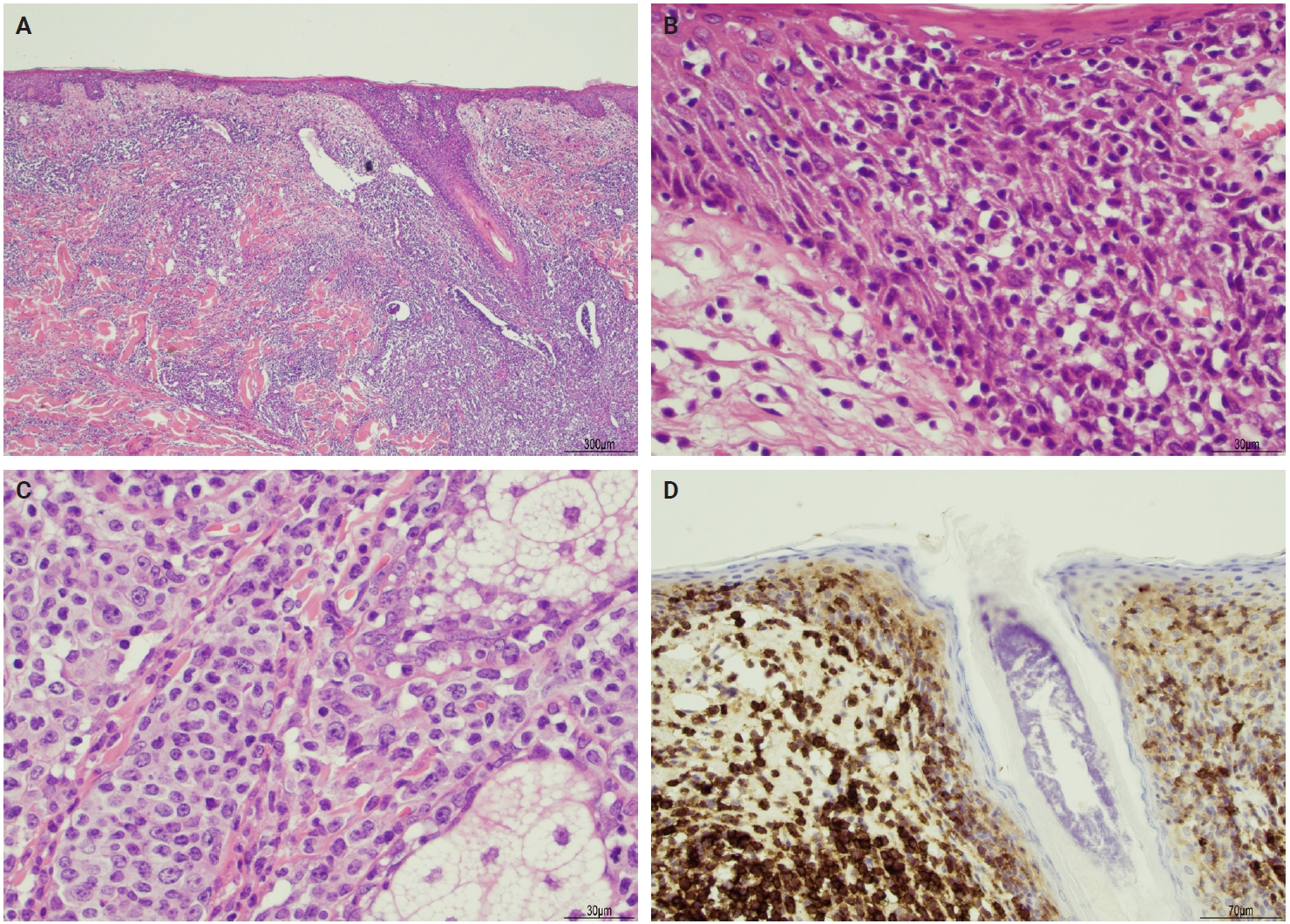
- The microscopic architecture and immunophenotype of large cells and their background are variable and have led to subclassification of LyP into different categories [1,5,6,13,14,27-33]. In LyP type A, neoplastic CD30+ cells are scattered and admixed with an extensive lymphoid infiltrate of neutrophils, eosinophils, and histiocytes mimicking classic Hodgkin lymphoma (Fig. 2B). In LyP type B, the presentation is characterized by epidermotropism and band-like distribution of small to medium atypical lymphocytes with cerebriform nuclei, usually CD30-negative, and mimicking MF, patch stage. Type C of LyP is composed of sheets of large neoplastic cells, with or without epidermotropism that display uniform positivity for CD30, mimicking a pc-ALCL; in this type, only few admixed inflammatory cells are noted. (Fig. 2C). In contrast, LyP type D presents with atypical small to medium-sized lymphocytes with distinct epidermotropism and these neoplastic cells express CD30 and CD8 and mimic pagetoid reticulosis. LyP type E shows an angiocentric and angiodestructive pleomorphic lymphoid infiltrate of small- to medium-sized lymphocytes admixed with scattered large cells uniformly positive for CD30 and the overall appearance is that of an aggressive lymphoma, such as extranodal T/natural killer (NK) cell lymphoma (Fig. 2D). LyP with DUSP22/IRF4 rearrangement (please note that it is not DUSP22::IRF4 since there is not a rearrangement between DUSP22 and IRF4; they are genomically too close for fluorescence in situ hybridization to separate them, so if positive, the rearrangement can be for DUSP22 or IRF4) displays a dense diffuse lymphoid infiltrate (Fig. 3A). A biphasic growth pattern with pagetoid reticulosis-like epidermotropism of small to medium size cerebriform lymphocytes (Fig. 3B) that express dim or lack CD30, while the second component is dermal or periadnexal (Fig. 3C) and the atypical lymphocytes express bright CD30+ (Fig. 3D) [25].
- Rare forms of LyP presentation include folliculotropic, syringotropic, granulomatous, and gamma-delta variant. In all cases, large atypical CD30+ cells are identified. In the folliculotropic pattern, there is a perifollicular infiltrate of atypical lymphocytes, cystic dilatation sometimes with rupture of the hair follicle, hyperplasia of the follicular epithelium, intrafollicular pustules (neutrophil collections), and follicular mucinosis [34]. The syringotropic pattern is characterized by eccrine units with periglandular infiltrate [14], while the granulomatous LyP shows mononuclear infiltrate with perivascular, eccrinotropic, and neurotropic distribution associated with non-caseating granulomas [14,35]. The gamma-delta variant may mimic aggressive lymphomas such as primary cutaneous gamma-delta T-cell lymphoma or MF. Most of the cases show cytotoxic immunophenotype; however, the clinical presentation is more akin to LyP [14,36]. Different histologic subtypes may coexist simultaneously or be present in the same patient throughout the clinical evolution of the lesions and even be associated with other CD30+ lymphoproliferative disorders such as pc-ALCL [37].
HISTOPATHOLOGICAL FINDINGS
- The neoplastic cells of LyP usually express CD3, CD4, CD25, CD30, CD45RO, human leukocyte antigen–DR isotype (HLA-DR), T-cell intracellular antigen-1 (TIA1), and granzyme B consistent with an activated T helper immunophenotype. A variable loss of T-cell antigens may be seen. In effect, these cells are characterized by the positivity for CD3 and CD4 as T helper cells. CD45RO defines them as memory/activated T cells while CD25, CD30, and HLA-DR define them as activated lymphocytes. Lastly, TIA1 and granzyme B indicate their cytotoxic phenotype [5,6,24,27-30]. Other T-cell antigens, such as CD2, CD5, and CD7, have a variable expression including complete negativity, depending on the subtype of LyP. Although CD8 is negative in LyP types A, B, and C, it is usually positive in LyP type D and has a variable positivity in type E and DUSP22/IRF4 rearrangement cases. The biomarker scenario of LyP is mainly characterized by the CD30 expression in the neoplastic cells, helping not only in the diagnostic definition of the entity, but also with the possibilities of new therapeutic options. Table 1 summarizes the main immunophenotypical features of different subtypes of LyP.
- Monoclonal rearrangements of the T-cell receptor gamma (TRG) or beta (TRB) chains are usually detected [5] and may overlap with other coexisting lymphomas, such as MF and pc-ALCL [38]. Recurrent mutations in epigenetic modifying genes (SETD2, KMT2A, KMT2D, and CREBBP) are the most frequently seen, but no specific mutations are observed, since a similar mutational profile is seen in pc-ALCL [39]. Recurrent NPM1::TYK2 gene fusions in LyP induce activation of the STAT signaling pathway [40,41]. More recently, cases with characteristic DUSP22/IRF4 rearrangement/6p25.3 translocation cases have been described and show characteristic features, such as common presentation in older patients, localized lesions with clinical presentation suggesting benign inflammatory dermatoses or epithelial tumors, and histological presentation with a bimodal pattern. The epidermotropic lymphocytes are small and are negative or express weak CD30, while the dermal cells are large, express strong CD30 and lymphocyte enhancer binding-factor 1, and are negative for TIA1 [6].
IMMUNOHISTOCHEMICAL AND ANCILLARY STUDIES
- The differential diagnosis of LyP is broad due to the great overlap in the clinical, histological, and immunophenotypical presentations with malignant skin diseases as well as benign dermatoses. The main differential diagnoses according to the specific type of LyP are summarized in Tables 2 and 3.
- Primary cutaneous anaplastic large cell lymphoma (pc-ALCL)
- Pc-ALCL is the main differential diagnosis of LyP type A and type C [27,42]. Both LyP and pc-ALCL are more prevalent in young adults. In LyP, the clinical presentation is characterized by multiple papules, papulonodular or nodular lesions <2 cm, while in pc-ALCL localized or single nodules or papules more than 2 cm are more common. The histological presentation in pc-ALCL is characterized by diffuse and homogeneous dermal infiltration of anaplastic cells without epidermotropism, while in LyP could vary from polymorphous and wedge-shaped in LyP type A and sheets of large lymphocytes with or without epidermotropism with few admixed inflammatory cells in type C. Cases of LyP type C could be morphologically identical to pc-ALCL, and because of this, the clinical correlation is fundamental for the final diagnosis. Both entities have a T-helper immunophenotype, characterized by CD3+ and CD4+, negativity for CD8, and diffuse positivity for CD30. Cases of pc-ALCL can also harbor DUSP22/IRF4 gene rearrangements, while cases of LyP usually express multiple myeloma oncogene 1/IRF4 and are negative for anaplastic lymphoma kinase-1, being additional markers that could help in the differential diagnosis [43,44]. The outcome is excellent in both entities [27,42].
- Early-stage MF
- Cases of LyP type B could have some overlapping histological and immunophenotypical presentation with early-stage MF. Both neoplasms present in adults and have a slight predominance in males. The clinical presentation in LyP is characterized by papular, papulonodular, or nodular lesions, while early-stage MF presents with patches and plaques. Histologically, an epidermotropic infiltrate of small/medium cerebriform cells is observed in both entities and might be indistinguishable. The immunophenotype is also very similar in both, being CD3+, CD4+, CD8–, and CD30–. The clinical correlation is fundamental to precisely determine the diagnosis [27,45].
- Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma
- This is a rare and clinically aggressive primary cutaneous lymphoma with a poor prognosis, which may have overlapping features with LyP type D. Both affect young adult men; however, the clinical presentation of primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma (PC CD8-AECTCL) is more dramatic and the lesions are usually ulcerative patches or plaques with rapid progression and necrosis. The main immunophenotypic difference is centered on the CD30 expression, which is positive in LyP and negative in PC CD8-AECTCL; however, the cytotoxic immunophenotype (CD3+CD4–CD8+) is common in both [30,46].
- Extranodal NK/T-cell lymphoma involving skin
- LyP type E is the main differential diagnosis of extranodal NK/T-cell lymphoma (ENKTCL). Both affect more men in the fifth to sixth decade of life. While LyP type E presents with recurrent clustered papular lesions with rapid progression to hemorrhagic necrotic ulcers, ENKTCL presents with multiple necrotic lesions. The histological presentation can be identical in both, characterized by angiocentric and angiodestructive pleomorphic lymphoid infiltrate of small to medium size and significant inflammatory background. The main immunophenotypical difference is centered on the association with Epstein-Barr virus, which is present in ENKTCL and negative in LyP type E. ENKTCL is also very aggressive and has a bad prognosis [6,27,47].
- Large cell transformation of MF
- Both LyP with DUSP22/IRF4 rearrangement and large cell transformation of MF affect elderly men. LyP with DUSP22/IRF4 presents with one or multiple eruptive and papulonodular lesions in a single body area, while patients with large cell transformation of MF refer to a history of evolution of patches, plaques, and tumors. This LyP subtype usually displays a biphasic growth pattern, where the epidermotropic component is that of small cells and the dermal/periadnexal component is composed of medium to large cells. Large cell transformation of MF has sheets of large cells, occasionally admixed with cerebriform lymphocytes. The immunophenotype is similar in both entities and CD30 tends to be more uniform in cases of LyP, but a diffuse and strong positivity cannot exclude large cell transformation of MF. The clinical correlation is fundamental to rendering the final diagnosis [6,48,49].
- Additional differential diagnosis
- Due to the broad histological and clinical manifestation of LyP, other entities that could also be considered as differential diagnosis, but will not be contemplated in this review, are (1) pityriasis lichenoides/pityriasis lichenoides et varioliformis acuta; (2) arthropod bite reaction; (3) reactive CD30+ cutaneous infiltrates; (4) systemic ALCL involving skin; and (5) classic Hodgkin lymphoma involving skin.
DIFFERENTIAL DIAGNOSIS
- Most patients do not require specific treatment [1,6,7,18], and clinical observation only is enough. Most of the treatments are aimed at symptom relief and faster regression. There are several treatment modalities, such as (1) low-dose methotrexate [11,18,31]; (2) psoralen ultraviolet A [1,7,11,18], which is related to longer disease-free survival: 10–36 months [18]; (3) topical corticosteroids [1,7,18]; (4) brentuximab vedotin, an active and well tolerated option and has shown a very high response rate in some reports [1,4]; (5) bexarotene [1]; (6) topical ruxolitinib [41]; and (7) topical alkylating agents (e.g., chlormethine gel) [50]. Treatment discontinuation often led to relapses and therapies often suppress lesions without altering the long-term disease course. Since the clinical course and the low necessity of treatment, few innovative therapeutic options are studied in clinical trials (https://clinicaltrials.gov/search?cond=Lymphomatoid%20Papulosis&viewType=Card).
- The prognosis is excellent [5,7,11,13], and only around 5% of the patients die of disease over a median period of 4 years, particularly patients with established underlying cutaneous lymphoma [1]. Although it is important to differentiate different subtypes of LyP for a proper differential diagnosis, the histologic types do not have prognostic significance [27].
- Patients with a diagnosis of LyP are recommended clinical follow-up, since there is a risk of developing a second lymphoid malignancy (e.g., MF and Hodgkin lymphoma) [4,5,7,11,13,18]. Patients with monoclonal TRB or TRG gene rearrangements or a mixed type of LyP have a higher risk of progressing to lymphoma [13], and an increased risk of subsequent lymphoma has been reported in men and in patients with LyP types B and C, while LyP type A and D are less frequently associated with lymphoma development [1]. Cases of LyP type A are the most prevalent subtype associated with early relapses [18] and elevated serum lactate dehydrogenase is associated with a high risk of associated lymphoproliferative disorders in all cases of LyP [8].
TREATMENT AND PROGNOSTIC FACTORS
Ethics Statement
Not applicable.
Availability of Data and Material
Data sharing not applicable to this article as no datasets were generated or analyzed during the study.
Code Availability
Not applicable.
Author Contributions
Writing—original draft: MLMP, CATC, RNM. Writing—review & editing: MLMP, CATC, RNM. Approval of final manuscript: all authors.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.
- 1. Wieser I, Oh CW, Talpur R, Duvic M. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol 2016; 74: 59-67. ArticlePubMed
- 2. Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia 2022; 36: 1720-48.
- 3. Wieser I, Wohlmuth C, Nunez CA, Duvic M. Lymphomatoid papulosis in children and adolescents: a systematic review. Am J Clin Dermatol 2016; 17: 319-27. ArticlePubMedPDF
- 4. Duvic M, Tetzlaff MT, Gangar P, Clos AL, Sui D, Talpur R. Results of a phase II trial of brentuximab vedotin for CD30+ cutaneous T-cell lymphoma and lymphomatoid papulosis. J Clin Oncol 2015; 33: 3759-65. ArticlePubMedPMC
- 5. Kempf W. A new era for cutaneous CD30-positive T-cell lymphoproliferative disorders. Semin Diagn Pathol 2017; 34: 22-35. ArticlePubMed
- 6. Karai LJ, Kadin ME, Hsi ED, et al. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid papulosis. Am J Surg Pathol 2013; 37: 1173-81. ArticlePubMed
- 7. Bekkenk MW, Geelen FA, van Voorst Vader PC, et al. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood 2000; 95: 3653-61. ArticlePubMedPDF
- 8. Lee WJ, Yun SJ, Jung JM, et al. Primary cutaneous CD30+ lymphoproliferative disorders in South Korea: a nationwide, multi-center, retrospective, clinical, and prognostic study. Ann Dermatol 2025; 37: 75-85. ArticlePubMedPMCPDF
- 9. Campo E, Jaffe ES, Cook JR, et al. The International Consensus Classification of Mature Lymphoid Neoplasms: a report from the Clinical Advisory Committee. Blood 2022; 140: 1229-53. ArticlePubMedPMC
- 10. Macaulay WL. Lymphomatoid papulosis: a continuing self-healing eruption, clinically benign: histologically malignant. Arch Dermatol 1968; 97: 23-30. ArticlePubMed
- 11. Kempf W, Zimmermann AK, Mitteldorf C. Cutaneous lymphomas: an update 2019. Hematol Oncol 2019; 37 Suppl 1: 43-7. ArticlePubMedPDF
- 12. Wang HH, Lach L, Kadin ME. Epidemiology of lymphomatoid papulosis. Cancer 1992; 70: 2951-7. ArticlePubMed
- 13. de Souza A, el-Azhary RA, Camilleri MJ, Wada DA, Appert DL, Gibson LE. In search of prognostic indicators for lymphomatoid papulosis: a retrospective study of 123 patients. J Am Acad Dermatol 2012; 66: 928-37. ArticlePubMed
- 14. Fricke T, Kempf W, Schon MP, Mitteldorf C. Histologic and immunohistochemical patterns in lymphomatoid papulosis: a systematic review of published cases. Dermatopathology (Basel) 2025; 12: 6.ArticlePubMedPMC
- 15. Blanchard M, Morren MA, Busschots AM, et al. Paediatric-onset lymphomatoid papulosis: results of a multicentre retrospective cohort study on behalf of the EORTC Cutaneous Lymphoma Tumours Group (CLTG). Br J Dermatol 2024; 191: 233-42. ArticlePubMedPDF
- 16. Friedman A. JAAD Game Changer: Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol 2025; 92: 251.ArticlePubMed
- 17. Moura FN, Thomas L, Balme B, Dalle S. Dermoscopy of lymphomatoid papulosis. Arch Dermatol 2009; 145: 966-7. ArticlePubMed
- 18. Fernandez-de-Misa R, Hernandez-Machin B, Servitje O, et al. First-line treatment in lymphomatoid papulosis: a retrospective multicentre study. Clin Exp Dermatol 2018; 43: 137-43. ArticlePubMedPDF
- 19. Nakajima I, Takeuchi K, Tsuji H. Ulcerated eyelid lesion leading to a lymphomatoid papulosis diagnosis. J Fr Ophtalmol 2025; 48: 104395.ArticlePubMed
- 20. de-Misa RF, Garcia M, Dorta S, et al. Solitary oral ulceration as the first appearance of lymphomatoid papulosis: a diagnostic challenge. Clin Exp Dermatol 2010; 35: 165-8. ArticlePubMed
- 21. Brown JR, Skarin AT. Clinical mimics of lymphoma. Oncologist 2004; 9: 406-16. ArticlePubMedPDF
- 22. Min JA, Oh ST, Kim JE, Cho BK, Chung NG, Park HJ. Lymphomatoid papulosis followed by anaplastic large cell lymphoma in a pediatric patient. Ann Dermatol 2010; 22: 447-51. ArticlePubMedPMC
- 23. Caccavale S, Vitiello P, Mascolo M, Ciancia G, Argenziano G. Dermoscopy of different stages of lymphomatoid papulosis. J Eur Acad Dermatol Venereol 2018; 32: e198-200. ArticlePubMedPDF
- 24. Namasondhi A, Rutnin S, Jerasutus S, Boonsakan P, Triyangkulsri K. Lymphomatoid papulosis type E with T-cell receptor gamma positivity. Clin Cosmet Investig Dermatol 2025; 18: 177-82. ArticlePubMedPMCPDF
- 25. Torres-Cabala CA. Diagnosis of T-cell lymphoid proliferations of the skin: putting all the pieces together. Mod Pathol 2020; 33: 83-95. ArticlePubMedPDF
- 26. Scarisbrick JJ, Calonje E, Orchard G, Child FJ, Russell-Jones R. Pseudocarcinomatous change in lymphomatoid papulosis and primary cutaneous CD30+ lymphoma: a clinicopathologic and immunohistochemical study of 6 patients. J Am Acad Dermatol 2001; 44: 239-47. ArticlePubMed
- 27. Willemze R, Cerroni L, Kempf W, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019; 133: 1703-14. ArticlePubMedPMCPDF
- 28. Kempf W, Kazakov DV, Baumgartner HP, Kutzner H. Follicular lymphomatoid papulosis revisited: a study of 11 cases, with new histopathological findings. J Am Acad Dermatol 2013; 68: 809-16. ArticlePubMed
- 29. Crowson AN, Baschinsky DY, Kovatich A, Magro C. Granulomatous eccrinotropic lymphomatoid papulosis. Am J Clin Pathol 2003; 119: 731-9. ArticlePubMed
- 30. Saggini A, Gulia A, Argenyi Z, et al. A variant of lymphomatoid papulosis simulating primary cutaneous aggressive epidermotropic CD8+ cytotoxic T-cell lymphoma: description of 9 cases. Am J Surg Pathol 2010; 34: 1168-75. ArticlePubMed
- 31. Calcaterra V, Cavalli R, Croci GA, et al. Type D lymphomatoid papulosis with pityriasis lichenoides et varioliformis acuta-like features in a child with parvovirus infection: a controversial diagnosis in the spectrum of lymphoid proliferations: case report and literature review. Ital J Pediatr 2022; 48: 183.ArticlePubMedPDF
- 32. Verheyden MJ, Venning VL, Khurana S, Cheung K. Follicular lymphomatoid papulosis - not a simple folliculitis. Australas J Dermatol 2021; 62: 235-7. ArticlePubMedPDF
- 33. Mark E, Kempf W, Guitart J, et al. Lymphomatoid papulosis with T-cell receptor-gamma delta expression: a clinicopathologic case-series of 26 patients of an underrecognized immunophenotypic variant of lymphomatoid papulosis. Am J Surg Pathol 2024; 48: 501-10. ArticlePubMed
- 34. Li X, Cheng S, Yu C, et al. Co-delivery of retinoic acid and miRNA by functional Au nanoparticles for improved survival and CT imaging tracking of MSCs in pulmonary fibrosis therapy. Asian J Pharm Sci 2024; 19: 100944.ArticlePubMedPMC
- 35. Slone SP, Martin AW, Wellhausen SR, et al. IL-4 production by CD8+ lymphomatoid papulosis, type C, attracts background eosinophils. J Cutan Pathol 2008; 35 Suppl 1: 38-45. ArticlePubMed
- 36. Weiner D, Carter JB, Lansigan F, LeBlanc RE. Primary cutaneous CD30-positive lymphoproliferative disorder with gamma-delta T-cells: a molecular-annotated case with a classic clinical appearance and behavior. J Cutan Pathol 2026; 53: 318-22. ArticlePubMedPDF
- 37. Goodlad JR, Cerroni L, Swerdlow SH. Recent advances in cutaneous lymphoma: implications for current and future classifications. Virchows Arch 2023; 482: 281-98. ArticlePubMedPMCPDF
- 38. de la Garza Bravo MM, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol 2015; 46: 558-69. ArticlePubMed
- 39. Abdulla FR, Zhang W, Wu X, et al. Genomic analysis of cutaneous CD30-positive lymphoproliferative disorders. JID Innov 2022; 2: 100068.ArticlePubMedPMC
- 40. Velusamy T, Kiel MJ, Sahasrabuddhe AA, et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood 2014; 124: 3768-71. ArticlePubMedPDF
- 41. Wobser M, Goebeler M, Rosenwald A, Maurus K. Topical ruxolitinib for treatment of mycosis fungoides and lymphomatoid papulosis. Br J Dermatol 2025; 193: 563-4. ArticlePubMedPDF
- 42. Magro CM, Momtahen S, Kiuru M. Primary cutaneous small cell variant of anaplastic large cell lymphoma: a case series and review of the literature. Am J Dermatopathol 2017; 39: 877-89. ArticlePubMed
- 43. Kempf W, Kutzner H, Cozzio A, et al. MUM1 expression in cutaneous CD30+ lymphoproliferative disorders: a valuable tool for the distinction between lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Br J Dermatol 2008; 158: 1280-7. ArticlePubMed
- 44. Onaindia A, de Villambrosía SG, Prieto-Torres L, et al. DUSP22-rearranged anaplastic lymphomas are characterized by specific morphological features and a lack of cytotoxic and JAK/STAT surrogate markers. Haematologica 2019; 104: e158-62. ArticlePubMedPMC
- 45. Amorim GM, Niemeyer-Corbellini JP, Quintella DC, Cuzzi T, Ramos-E-Silva M. Clinical and epidemiological profile of patients with early stage mycosis fungoides. An Bras Dermatol 2018; 93: 546-52. ArticlePubMedPMC
- 46. Guitart J, Martinez-Escala ME, Subtil A, et al. Primary cutaneous aggressive epidermotropic cytotoxic T-cell lymphomas: reappraisal of a provisional entity in the 2016 WHO classification of cutaneous lymphomas. Mod Pathol 2017; 30: 761-72. ArticlePubMedPMCPDF
- 47. Haverkos BM, Pan Z, Gru AA, et al. Extranodal NK/T cell lymphoma, nasal type (ENKTL-NT): an update on epidemiology, clinical presentation, and natural history in North American and European cases. Curr Hematol Malig Rep 2016; 11: 514-27. ArticlePubMedPMCPDF
- 48. Feldman AL, Oishi N, Ketterling RP, Ansell SM, Shi M, Dasari S. Immunohistochemical approach to genetic subtyping of anaplastic large cell lymphoma. Am J Surg Pathol 2022; 46: 1490-9. ArticlePubMedPMC
- 49. Pulitzer M, Myskowski PL, Horwitz SM, et al. Mycosis fungoides with large cell transformation: clinicopathological features and prognostic factors. Pathology 2014; 46: 610-6. ArticlePubMedPMC
- 50. Dege T, Netzer I, Strobel K, Goebeler M, Wobser M. Successful treatment of lymphomatoid papulosis with chlormethine gel. J Dtsch Dermatol Ges 2024; 22: 826-7. Article
REFERENCES
Figure & Data
References
Citations
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
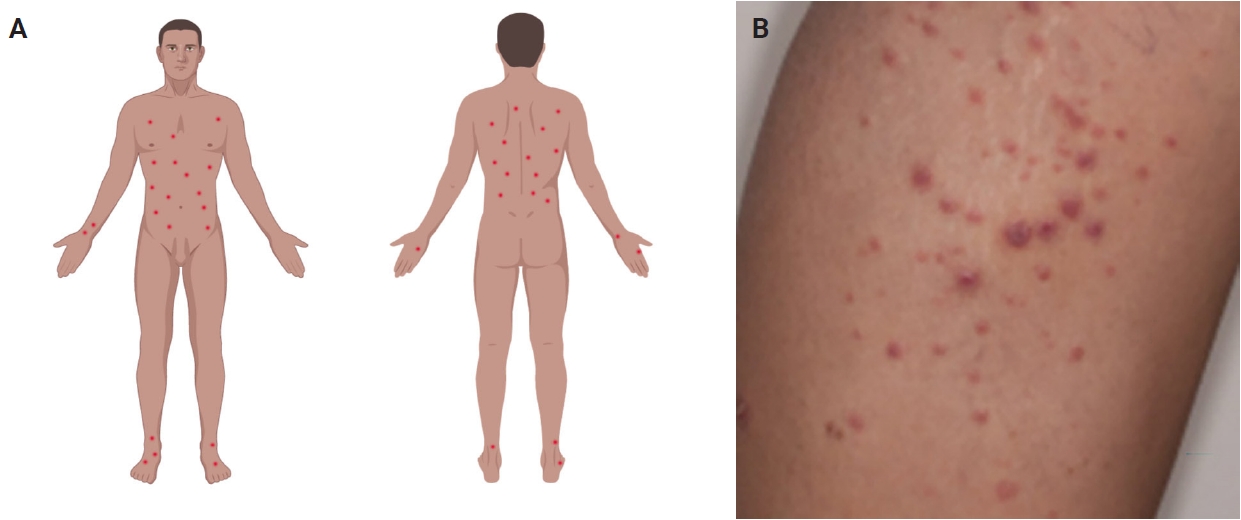
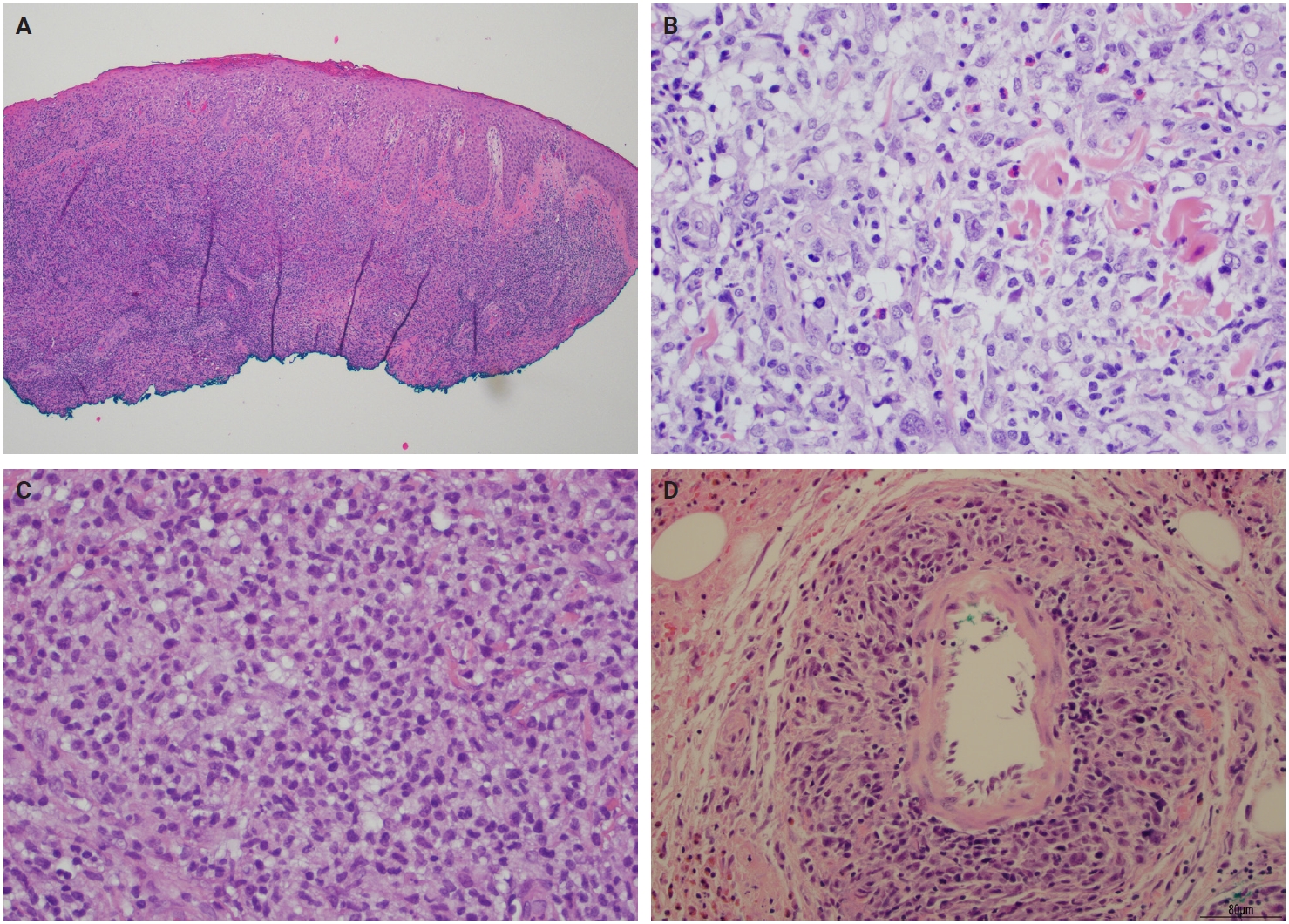
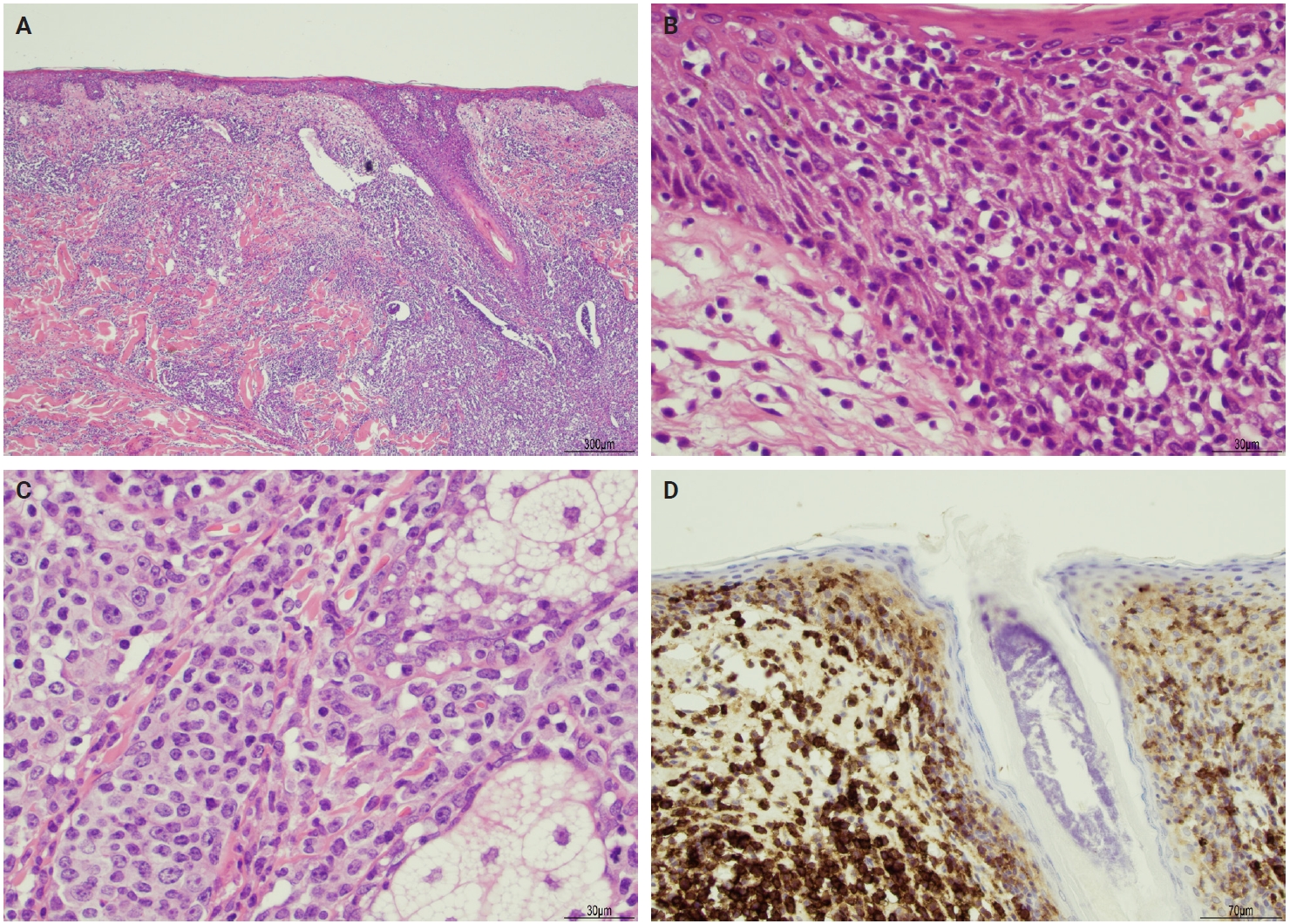
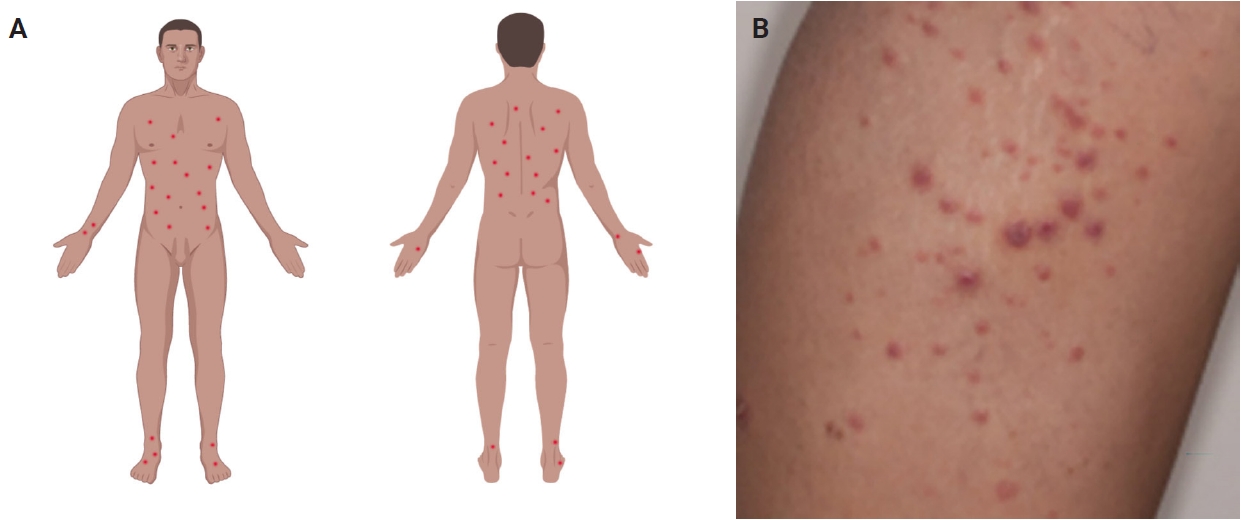

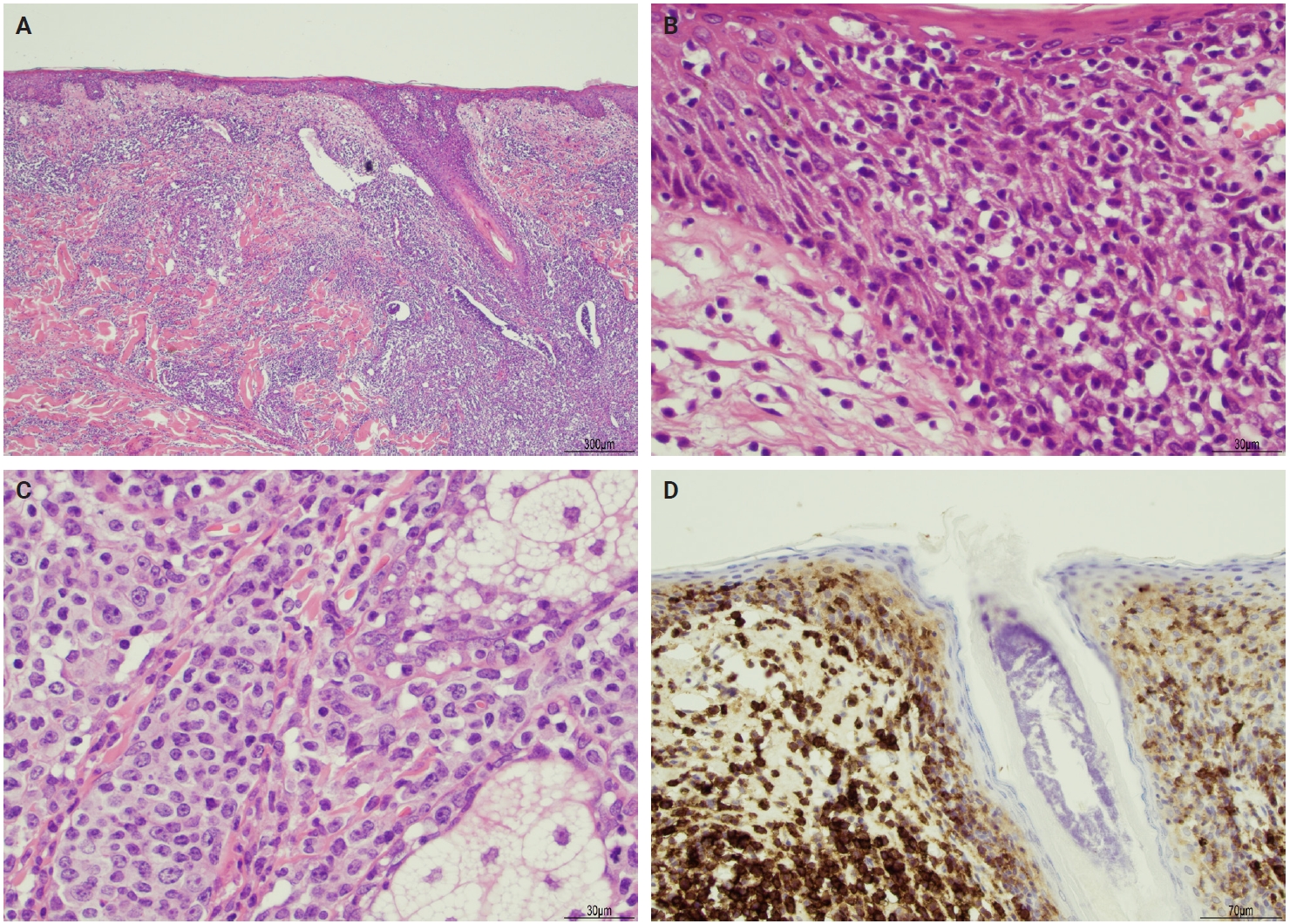
Fig. 1.
Fig. 2.
Fig. 3.
| Type of LyP | CD2 | CD3 | CD4 | CD5 | CD7 | CD8 | CD30 | ALK1 | Other markers |
|---|---|---|---|---|---|---|---|---|---|
| Type A | +/– | + | + | +/– | +/– | – | + | – | TIA1+ |
| Type B | +/– | + | + | +/– | +/– | – | – | – | TIA1+ (expression variable) |
| Type C | +/– | + | + | +/– | +/– | – | + | – | TIA1+ |
| Type D | + | + | – | – | – | + | + | – | TIA1+ |
| βF1+ | |||||||||
| CD56– | |||||||||
| EBER– | |||||||||
| Type E | + | + | –/+ | + | +/– | + | + | – | CD45RA– |
| CD45RO+/– | |||||||||
| TIA1+ | |||||||||
| βF1+ | |||||||||
| EBER– | |||||||||
| LyP with DUSP22/IRF4 rearrangement | +/– | + | – | +/– | +/– | +/– | + | – | CD15+, LEF1+ |
| TIA1– | |||||||||
| Ki67 (>80%) | |||||||||
| TCRβ+ | |||||||||
| Folliculotropic LyP | + | + | + | +/– | – | – | + | – | - |
| Granulomatous LyP | + | + | + | + | – | – | + | – | - |
| Age | Sex (M:F) | Clinical feature | Treatment | Outcome | |
|---|---|---|---|---|---|
| LyP type A | Fifth decade | 2–3:1 | Papular, papulonodular or nodular; <2 cm | None for spontaneous regression; symptomatic treatment for relief | Excellent |
| Primary cutaneous anaplastic large cell lymphoma | Seventh decade | 2–3:1 | Localized nodule or papule; usually >2 cm | Excision, radiation, or chemotherapy, alone or combined | Excellent |
| LyP type B | Fifth decade | 2–3:1 | Papular, papulonodular, or nodular | Spontaneous regression or symptomatic treatment | Excellent |
| Early-stage mycosis fungoides | Sixth decade | 2:1 | Patches and plaques | Ultraviolet light | Good |
| LyP type C | Fifth decade | 2–3:1 | Papular, papulonodular, or nodular; <2 cm | Spontaneous regression or symptomatic treatment | Excellent |
| Primary cutaneous anaplastic large cell lymphoma | Seventh decade | 2–3:1 | Localized nodule or papule; >2 cm | Radiation or chemotherapy | Good |
| LyP type D | Third decade | 2:1 | Clustered papules or small nodules | Spontaneous regression or symptomatic treatment | Excellent |
| Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma | Fourth decade | 5:2 | Ulcerative patches or plaques | Chemotherapy, stem cell transplant | Poor |
| LyP type E | Sixth decade | 3:1 | Recurrent clustered papular lesions with rapid progression to hemorrhagic necrotic ulcers | Spontaneous regression or symptomatic treatment | Excellent |
| Extranodal NK/T-cell lymphoma | Fifth and sixth decades | 2:1 | Multiple necrotic lesions | Progressive disease | Poor |
| LyP with DUSP22/IRF4 rearrangement | Eighth decade | 4.5:1 | 1 or multiple eruptive and papulonodular lesions in a single body area | Spontaneous regression or symptomatic treatment | Excellent |
| Transformed mycosis fungoides | Seventh decade | 1.1:1 | Patches, plaques, and tumor | Radiation, chemotherapy | Poor |
| Folliculotropic LyP | Sixth decade | 4.5:1 | Follicular pustules | Spontaneous regression | Excellent |
| Folliculitis | Variable | Variable | Superficial folliculitis | Topical treatment | Excellent |
| Granulomatous LyP | Fifth decade | 5:4 | Nodules | Spontaneous regression | Excellent |
| Discoid lupus erythematosus | Fifth decade | 1:2 | Discoid lesions | Systemic agents, phototherapy | Excellent |
| Microscopy | Immunophenotype | |
|---|---|---|
| LyP type A | Wedge-shaped (subtle) with scattered Hodgkin/Reed-Sternberg–like cells admixed with neutrophils, eosinophils, and histiocytes | CD3+, CD4+, CD8–, CD30+ |
| Primary cutaneous anaplastic large cell lymphoma | Diffuse and homogeneous dermal infiltrate of anaplastic large cells | CD3 (variable), CD4+, CD8–, CD30+ |
| LyP type B | Epidermotropism and band-like distribution of small/medium atypical lymphocytes with cerebriform nuclei (MF-like) | CD4+, CD8–, CD30– |
| Early-stage MF | Cerebriform cells with epidermotropism predominate along the basal layer of epidermis | CD4+, CD7–, CD8–, CD30– |
| LyP type C | Sheets of large lymphocytes with or without epidermotropism and few admixed inflammatory cells | CD4+, CD8–, CD30+ (uneven) |
| Primary cutaneous anaplastic large cell lymphoma | Diffuse dermal infiltrate of large anaplastic lymphocytes | CD3 (variable), CD4+, CD8–, CD30+ (uniform) |
| LyP type D | Epidermotropism with pagetoid reticulosis-like | CD2+, CD3+, CD4–, CD8+, CD30+, TIA1+ |
| Primary cutaneous CD8+ aggressive epidermotropic cytotoxic T-cell lymphoma | Small to intermediate-sized pleomorphic lymphocytes with epidermotropism | CD2–, CD3+, CD7+, CD8+, CD30–, CD56– |
| LyP type E | Angiocentric and angiodestructive pleomorphic lymphoid infiltrate of small/medium size | CD2+, CD3+, CD4–/+, CD8+, CD30+, TIA1+, EBV– |
| Extranodal NK/T-cell lymphoma | Atypical lymphocytes; angiocentric and angiodestructive in most cases | CD3–, CD4–, CD5–, CD8–/+, CD30+, CD56+, TIA1+, EBER+ |
| LyP with DUSP22/IRF4 rearrangement | Biphasic growth pattern: epidermotropic small cells, versus dermal or periadnexal medium to large cells | CD3+, CD4–, CD8 (variable), LEF1+, TIA1– |
| Transformed MF | Cerebriform with increased large cells in the dermis | CD3+, CD4+, CD7–, CD8–, CD30 (variable) |
| Folliculotropic LyP | Perifollicular infiltrate of atypical lymphocytes and follicular mucinosis | CD3+, CD4+, CD30+ |
| Folliculitis | Moderate mixed inflammatory cells in the follicular ostium | Polyclonal |
| Granulomatous LyP | Noncaseating granulomas associated with mononuclear infiltrate with perivascular, eccrinotropic, and neurotropic distribution | CD3+, CD4+, CD5–, CD7–, CD8–, CD30+ |
| Discoid lupus erythematosus | Edema and a mild infiltrate of inflammatory cells (lymphocytes) in the upper dermis | CD3+, CD4+; no loss of T-cell antigens |
LyP, lymphomatoid papulosis; ALK1, anaplastic lymphoma kinase 1; TIA1, T-cell intracellular antigen 1; EBER, Epstein-Barr virus-encoded small RNA; LEF1, lymphoid enhancer-binding factor 1; TCRβ, T-cell receptor beta chain.
LyP, lymphomatoid papulosis.
LyP, lymphomatoid papulosis; MF, mycosis fungoides; TIA1, T-cell intracellular antigen-1; NK, natural killer; EBV, Epstein-Barr virus; EBER, Epstein-Barr virus-encoded small RNA.

