Clear Cell Papillary Renal Cell Carcinoma: A Report of 15 Cases Including Three Cases of Concurrent Other-Type Renal Cell Carcinomas
Article information
Abstract
Background
Clear cell papillary renal cell carcinoma (CCPRCC) is a recently established subtype of renal epithelial tumor. The aim of this study was to identify the diagnostic criteria of CCPRCC with an emphasis on immunohistochemical studies, and to report three cases with concurrent other-type renal cell carcinoma (RCC).
Methods
A total of 515 RCC patients that consecutively underwent surgical resection at Seoul National University Hospital from 1 January 2010 to 31 December 2011 were screened. Each case was reviewed based on the histologic features and was evaluated immunohistochemically.
Results
A total of 15 CCPRCCs were identified, which composed 2.9% of the total RCCs. The mean age was 52 years, and the average tumor size was 1.65 cm. All 15 cases showed low nuclear grade, no lymph node metastasis and no distant metastasis. The CCPRCCs showed variable architectural patterns including cystic, trabecular, papillary, and acinar. All of the cases showed moderate to intense immunoreactivity for cytokeratin 7 (CK7). CD10 was negative or showed focal weak positivity. Three cases had concurrent other-type RCC, including a clear cell RCC and an acquired cystic disease-associated RCC.
Conclusions
The strong CK7 and negative or focal weak CD10 expression will be useful for the diagnosis of CCPRCC.
The 2004 World Health Organization (WHO) classification of renal epithelial tumors, which is the current one, is based on the histopathologic, immunohistochemical, and genetic features.1 As the knowledge of and experience with renal epithelial tumors have increased, new entities and morphologic variants of the main tumor categories have emerged2,3 including tubulocystic carcinoma, thyroid follicular carcinoma-like tumor, clear cell papillary renal cell carcinoma (CCPRCC), and oncocytic papillary renal cell carcinoma. A precise understanding and correct diagnosis of renal tumors are essential not only for the investigation of cancers but also for prognostic and therapeutic purposes.4-6
CCPRCC has been recently established as a new type of renal tumor.7-15 Initially, this tumor was known as renal cell carcinoma (RCC) occurring in association with end-stage renal disease (ESRD),7 but several studies have reported cases of CCPRCC in patients without ESRD.9,10,12,13 CCPRCC typically presents as a small renal tumor with an early stage. Histopathologically, CCPRCC shows a tubular and papillary architecture with variable amounts of clear cytoplasm, a low Fuhrman nuclear grade, and a characteristic nuclear arrangement away from the basement membrane.9,11,15 Additionally, immunohistochemical staining demonstrates strong expression of cytokerain 7 (CK7), no or low expression of CD10 and racemase and negative expression of transcription factor E3 (TFE3).9,10,14,15 Molecular genetic studies have revealed that CCPRCCs harbor different pathogenic mechanisms from clear cell RCCs (CCRCCs) and papillary RCCs (PRCCs).12-14,16 CCPRCCs show no loss of chromosome 3p where the von Hippel-Lindau (VHL) gene is located, which is associated with CCRCC. Additionally, no gains of chromosomes 7 or 17, and no chromosomal imbalance have been observed, which are characteristic features of PRCC.
In this study, we investigated the clinicopathologic features of 15 CCPRCCs. Further, we studied the immunophenotypic features of the CCPRCCs, and we have attempted to suggest approaches for the differential diagnosis of small RCCs with clear cytoplasms with an emphasis on immunohistochemical studies. We also report three cases of a concurrent occurrence of CCPRCC and other-type RCC.
MATERIALS AND METHODS
Screening and inclusion criteria
A total of 515 patients with RCC who underwent radical or partial nephrectomy between 1 January 2010 and 31 December 2011 at Seoul National University Hospital were included in this study. Each RCC case was re-evaluated with regard to the RCC subtype; with reference to the "2009 update on the classification of renal epithelial tumors in adults."2 We retrieved the CCPRCC cases based on the pathologic features listed in several articles.9,12-14 RCCs with a primarily tubulopapillary architecture, clear cytoplasm, fibrous capsules, and characteristic nuclear alignment were considered the microscopic features of CCPRCCs. For a more accurate and precise retrieval, we performed CK7 and CD10 immunohistochemical staining on all 515 RCC cases. The cases with the appropriate histopathologic features, strong CK7 expression, and negative or focal weak CD10 expression were included in this study.
Clinicopathologic evaluation
A total of 15 cases of CCPRCC were identified. Tumor staging was defined according to the 2010 tumor-lymph nodes-metastasis (TNM) classification system,17 and nuclear grading was reviewed as in Fuhrman et al.18 Patient age, gender, and ESRD association were evaluated. RCC recurrence or metastasis was determined according to the clinical and radiographic findings. In each tumor section, we evaluated the histologic features that are common in CCPRCC. This study was approved by the Institutional Review Board of Seoul National University Hospital.
Immunohistochemistry
The immunohistochemical staining was performed on each representative slide of the 15 CCPRCCs. We evaluated immunoreactivity for CK7 (1:300, Dako, Glostrup, Denmark), CD10 (ready-to-use, Novocastra, Newcastle, UK), alpha-methylacyl-CoA racemase (AMACR; 1:300, Dako), TFE3 (1:1,500, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and vimentin (VT; 1:500, Dako). Each slide was dewaxed and rehydrated in a graded series of alcohol solutions. Immunohistochemical staining was performed using the Bond-Max autostainer (Leica Microsystems, Bannockburn, IL, USA) for CK7, CD10, TFE3, and VT, and the Dako Autostainer Link 48 (Dako Corp., Carpintera, CA, USA) was used for AMACR. The binding of the primary antibody was detected using the Bond polymer refine detection kit (Leica Microsystems) or the Dako EnVision Flex Kit according to the manufacturers' instructions.
RESULTS
The clinical and pathological characteristics of the CCPRCC patients
A total of 15 patients who underwent surgical resection and were diagnosed with CCPRCC were analyzed in this study. The clinicopathological characteristics of the patients are summarized in Table 1. The patients included 4 men and 11 women. The mean age was 52 years (range, 35 to 70 years), and the mean tumor size was 1.65 cm (range, 0.6 to 4.0 cm). Lymph node metastasis and distant metastasis were not observed. All cases were pT1aNxM0, and thus stage I. Of the 15 cases, 8 (53.3%) showed Fuhrman nuclear grade 2, and the remaining 7 (46.7%) were Fuhrman nuclear grade 1. An association with ESRD was identified in 4 cases (26.7%). The mean follow-up duration was 15.6 months (range, 9 to 29 months), and there was no case with disease progression or metastasis. Lymph node dissection was not performed in any of the 15 cases, and was classified as pNx according to the 2010 TNM classification system.17 A representative gross photograph and microscopic findings are shown in Fig. 1.
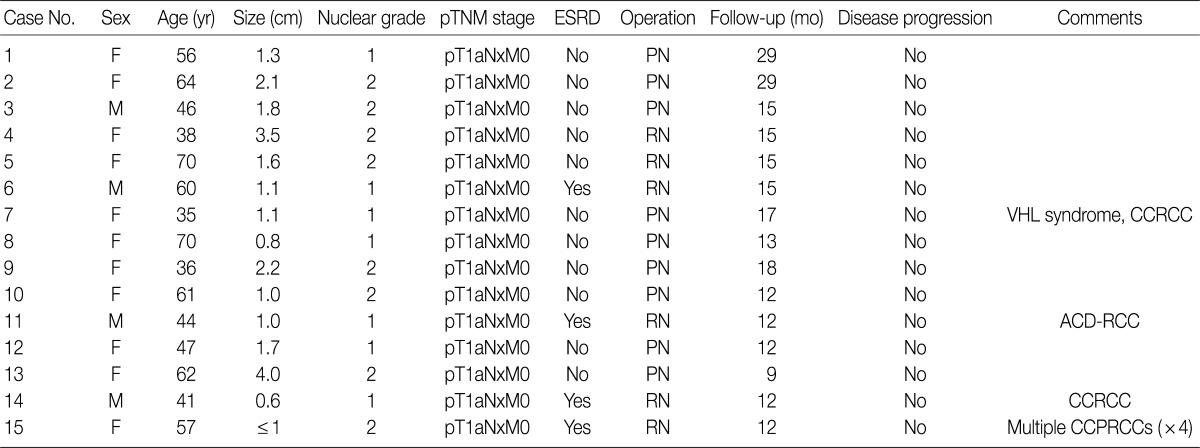
Clinicopathologic characteristics of the 15 CCPRCC patients
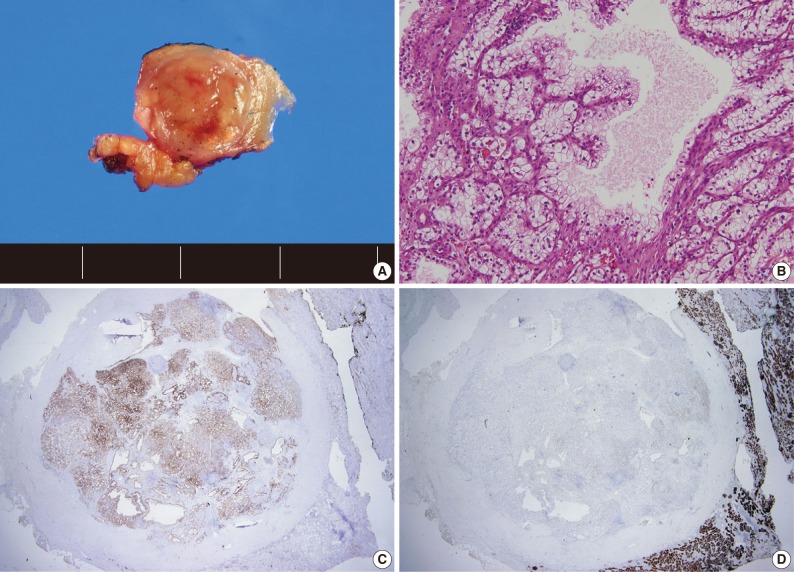
Representative gross photograph (A), microscopic findings (B), and immunohistochemical staining of cytokeratin 7 (C), and CD10 (D) of clear cell papillary renal cell carcinoma.
Multiple RCC cases
Four patients had multiple RCCs. Among them, 3 patients had CCPRCC with other-type RCC. Two patients had CCPRCC and concurrent CCRCC. One patient was clinically diagnosed with VHL syndrome (Fig. 2), and had a family history of VHL syndrome; the patient's father and brother were also affected. The patient underwent surgery for spinal capillary hemangioblastoma and had a history of a brain tumor. The patient also had a pancreatic cyst. Another patient had CCPRCC and a concurrent acquired cystic disease-associated RCC (ACD-RCC). The remaining patient showed four CCPRCCs in the same kidney and also suffered from ESRD.
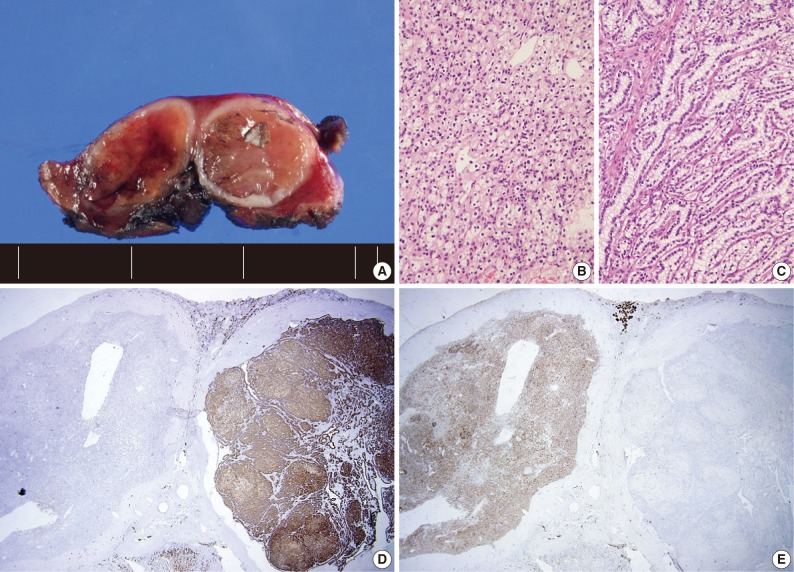
Case of the patient with von Hippel-Lindau syndrome with a concurrent occurrence of clear cell papillary renal cell carcinoma (CCPRCC) and clear cell renal cell carcinoma (CCRCC). A gross photograph (A) (left, CCRCC; right, CCPRCC), the microscopic findings of CCRCC (B) and CCPRCC (C), and the immunohistochemical staining of cytokeratin 7 (D) and CD10 (E) of both tumors.
Histopathologic features of the 15 CCPRCC cases
The architectural patterns (papillary, tubular, cystic, and acinar) were examined and are summarized in Table 2. We also evaluated the cytoplasmic clarity, characteristic nuclear arrangement, and encapsulation. These results are summarized in Table 3. A tubulopapillary architecture was identified in all 15 cases (100%), a cystic architecture in 14 cases (93.3%), and an acinar architecture in 9 cases (60.0%). All of the tumor cells were cuboidal with a low Fuhrman nuclear grade and clear cytoplasm. The characteristic nuclear alignment away from the basal aspect was identified in 8 cases (53.3%). All of the tumors were encapsulated by a fibrous capsule.
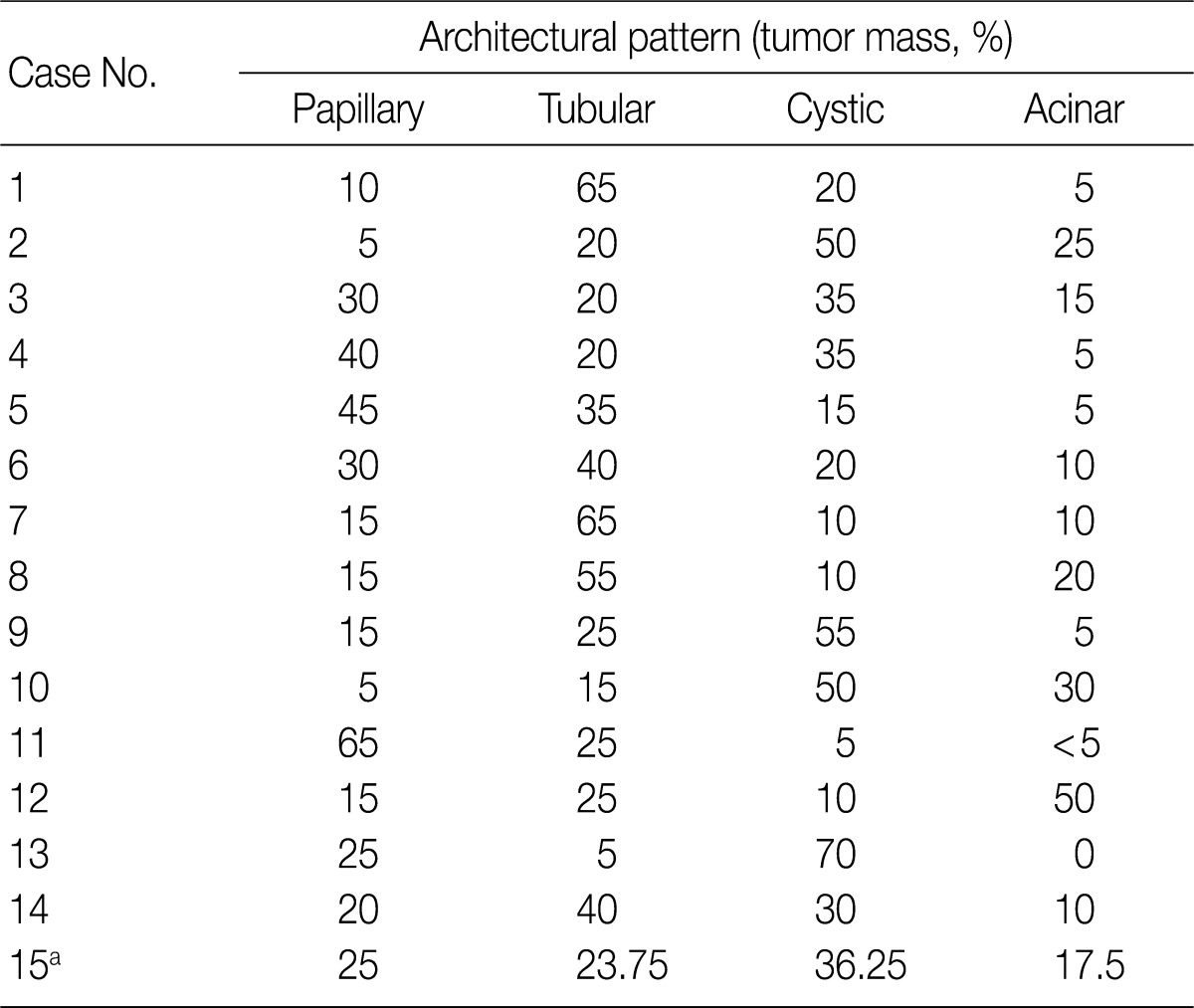
Histopathologic architectural patterns of the 15 CCPRCCs
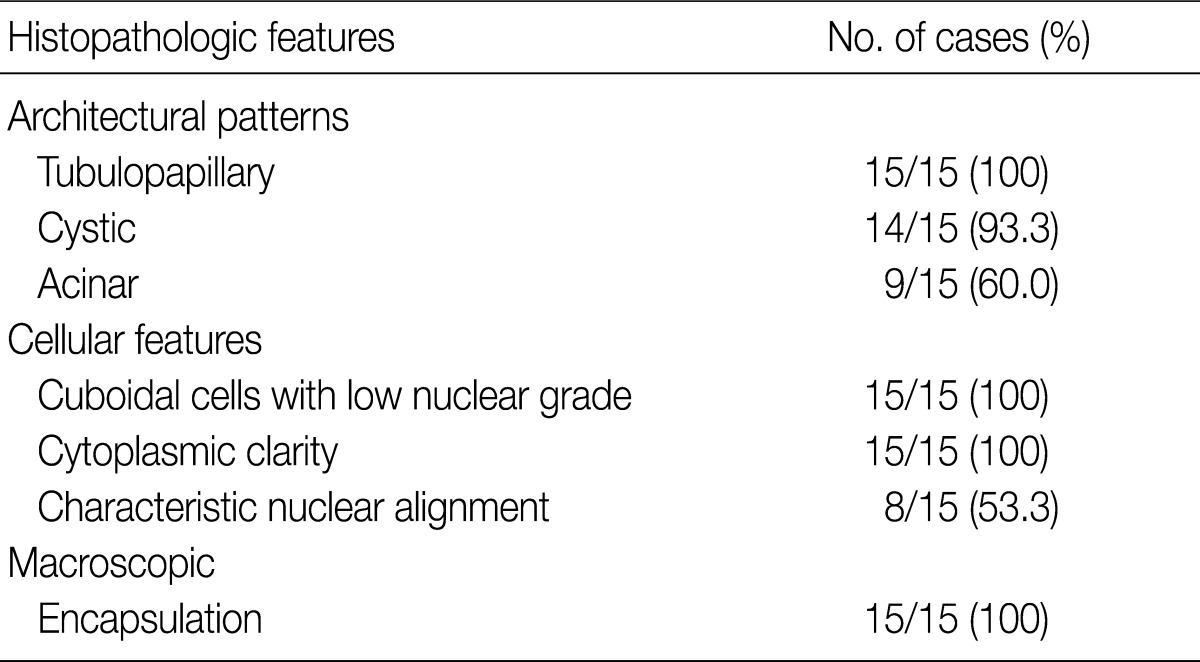
Histopathologic features of the 15 CCPRCCsa
Immunohistochemical features of the 15 CCPRCC cases
We assessed the immunohistochemical profile of CK7, CD10, AMACR, VT, and TFE3 in the 15 CCPRCC cases (Table 4). All 15 cases (100%) showed strong immunoreactivity for CK7. Eleven cases (73.3%) were negative for CD10, and 4 cases (26.7%) showed focal positivity. All of the cases were negative for AMACR and TFE3. All 15 cases were positive for VT immunoreactivity.
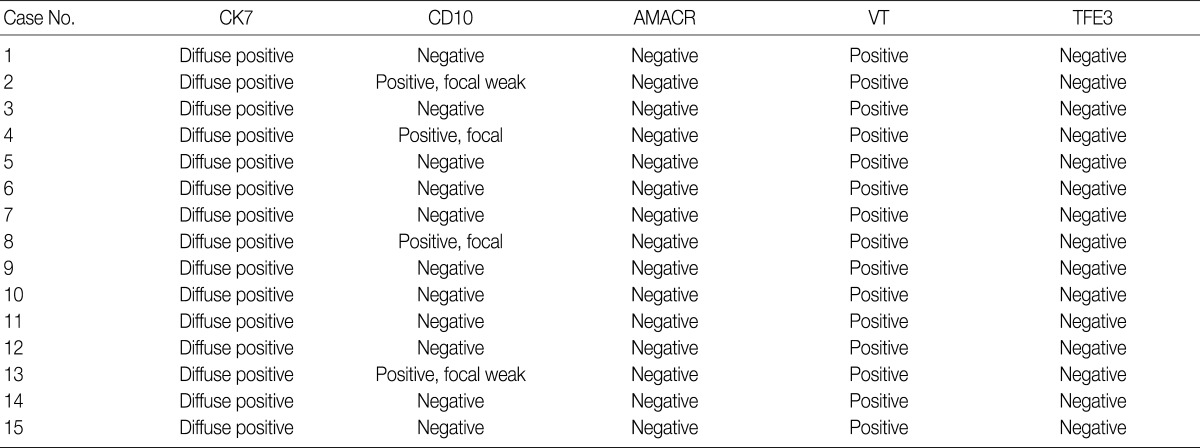
Immunohistochemical results of the 15 CCPRCCs
DISCUSSION
Due to the development of imaging modalities and the increased number of cases of early detection of RCC, the relative proportion of small RCCs has increased. Without an understanding of CCPRCC and its helpful diagnostic features, RCC can be misdiagnosed as CCRCC. Histopathologically, CCPRCC has shown a tubular and papillary architecture, tumor cells with clear cytoplasms, and a low Fuhrman nuclear grade; the characteristic nuclear arrangement away from the basement membrane has also been observed.9,11,15
CCPRCC was initially thought to occur in association with ESRD,7 and was regarded as a renal cell neoplasm in ESRD.2 However, several studies have reported that CCPRCC cases can occur in patients without ESRD.9,10,12,13 In our study, ESRD was associated with CCPRCC in only 4 cases (26.7%).
Most small CCRCCs show a low Fuhrman nuclear grade and display variable architectural patterns including solid, acinar, cystic, tubular, and papillary patterns.1,2 CCPRCCs can also show variable architectural patterns including tubulopapillary, solid, acinar, and cystic patterns.7-15 Moreover, the so-called characteristic nuclear arrangement of CCPRCC has not been identified in all cases and cannot be used to verify a diagnosis of CCPRCC. For instance, in our study, the characteristic nuclear alignment was identified in only 8 cases (53.3%). Therefore, a differential diagnosis between CCRCC and CCPRCC can be very difficult.
Renal carcinomas associated with Xp11.2 translocations are less commonly encountered in a differential diagnosis of CCPRCC.19 Typically, this entity presents at an advanced stage, and it is rarely detected in an early stage. Renal carcinomas associated with Xp11.2 translocations can show a papillary architecture and clear cells,1,19 and immunohistochemical staining for TFE3 and translocation studies can lead to a correct diagnosis. Papillary adenoma of the kidney would also be considered a differential diagnosis of CCPRCC. Papillary adenoma shows a papillary or tubular architecture and has a low nuclear grade. By definition, the diameter of a papillary adenoma is less than 5 mm. Though papillary adenoma has a similar architecture to CCPRCC and shows CK7 positivity, the tumor cells have features more similar to PRCC than CCPRCC, and the cytoplasm is not clear. These cytologic features may be helpful for the differential diagnosis of CCPRCC.
In our study, we identified the usefulness of immunohistochemical staining in the diagnosis of CCPRCC. All 15 CCPRCC cases showed diffuse positivity for CK7 and negative or focal weak positivity for CD10. In general, CCRCCs demonstrate strong and diffuse staining for CD10 and negative or focal weak staining for CK7,1,20 while CCPRCCs show the opposite results.9,10,14,15 Some histopathologic features, including a well-encapsulated mass, a primarily tubulopapillary architecture, and characteristic nuclear alignment, may increase the possibility of diagnosing CCPRCC, but these findings are somewhat subjective and do not always exist. However, CCRCCs can also show those histopathologic features. Therefore, a histologic examination may not be enough for the differential diagnosis between CCPRCC and CCRCC, and additional immunohistochemical staining for CK7 and CD10 may be very useful.
As far as we know, there have not been any reports of CCPRCC in Korea. Furthermore, there have been no reports on the proportion of CCPRCCs among all RCCs worldwide. In our study, we evaluated cases of surgically resected RCCs over a two-year period. The proportion of CCPRCCs among all of the RCCs was 2.9%. Compared with the frequency of chromophobe RCC, which accounts for approximately 5% of all RCCs,1 CCPRCC was not rare. Without an understanding of CCPRCC and without precise diagnostic histologic and immunohistochemical criteria, CCPRCCs can be misdiagnosed as CCRCCs.
We also assessed the prognosis of 15 CCPRCC cases. Though the mean follow-up duration was short (15.6 months), all 15 CCPRCC cases showed a favorable prognosis with no disease progression or no metastatis. These results were consistent with previous articles that have reported no recurrence or metastases in CCPRCC patients.7,9,12 There was one article that evaluated the prognosis of CCRCC according to pT staging.21 Though the article was based on the 5th American Joint Committee on Cancer (AJCC) cancer staging manual, and pT1 included tumors less than 7 cm; the 1 year cancer-specific survival rate of pT1 CCRCC was about 95% and 5 year survival rate was 88.7%.
Several reports have shown multiple CCPRCCs in the same patients. Gobbo et al.9 presented one patient with three CCPRCCs and ESRD. Aydin et al.12 included three patients with bilateral CCPRCC, and each patient had two CCPRCCs; the study also included one VHL patient with one CCPRCC. The study by Adam et al.13 contained four patients with multiple CCPRCCs; two patients had two bilateral CCPRCCs and impaired renal functions, and one patient had eight unilateral CCPRCCs and normal renal function. The fourth patient had multiple bilateral CCPRCCs and suffered from ESRD. Kuroda et al.16 reported three concurrent RCCs in a patient with impaired renal function. Among them, two tumors were CCRCCs, and one tumor was a CCPRCC.
In our cases, there were four multiple RCC cases. Interestingly, there were three cases that occurred with concurrent other-type RCC. One patient was clinically diagnosed with VHL syndrome with a family history of VHL syndrome (the patient's father and brother), capillary hemangioblastoma of the spine, and a brain tumor. As far as we know, this is the second CCPRCC case that has been reported to occur in a VHL syndrome patient. This patient showed normal renal function and had two RCCs of different histologic types, CCPRCC and CCRCC. A genetic study of the VHL gene mutation may show interesting results. However, we did not perform a genetic study of the VHL gene mutation in the CCRCC and CCPRCC cases in this study. We believe that this is a limitation of our study. Because the molecular pathogenesis of CCRCC and CCPRCC is considered to be different, investigating the VHL gene mutation status in both types of RCC may be interesting.
The second patient showed impaired renal function, and had ACD-RCC and CCPRCC. The third patient also showed impaired renal function, and had CCRCC and CCPRCC.
At the molecular level, CCPRCC showed different genetic alterations from CCRCC and PRCC.12-14,16 The loss of chromosome 3p, where the VHL gene and polybromo-1 (PBRM1) gene are located, is associated with CCRCC.1,22 Another genetic change identified in CCRCC is the loss of 9p and 14q.2 In PRCC cases, trisomy or tetrasomy of chromosome 7, trisomy 17, and a loss of the Y chromosome are characteristic features.23,24 However, CCPRCC does not have those genetic alterations.12-14,16 In CCPRCC, chromosomal alterations have been reported in a few cases. Aydin et al.12 reported low copy number gains at chromosomes 7 and 17 in one case of CCPRCC among 36 cases. Additionally, Kuroda et al.16 found polysomy for chromosome 7 and monosomy for chromosomes 17, 16, and 20 in one CCPRCC case. These results showed that CCPRCC is an entity unique from CCRCC and PRCC.
In summary, CCPRCC is a recently established type of renal epithelial tumor and one of the primary differential diagnoses of small RCCs with clear cytoplasms. The observation of strong CK7 expression and a negative or weak CD10 expression pattern is very useful for the differential diagnosis between CCRCC and CCPRCC.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (grant no. 2010-0004550).
Notes
No potential conflict of interest relevant to this article was reported.