 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 46(6); 2012 > Article
-
Original Article
Clear Cell Papillary Renal Cell Carcinoma: A Report of 15 Cases Including Three Cases of Concurrent Other-Type Renal Cell Carcinomas - Jeong Hwan Park1, Cheol Lee1, Ja Hee Suh1, Kyung Chul Moon1,2
-
Korean Journal of Pathology 2012;46(6):541-547.
DOI: https://doi.org/10.4132/KoreanJPathol.2012.46.6.541
Published online: December 26, 2012
1Department of Pathology, Seoul National University College of Medicine, Seoul, Korea.
2Kidney Research Institute, Medical Research Center, Seoul National University College of Medicine, Seoul, Korea.
- Corresponding Author: Kyung Chul Moon, M.D. Department of Pathology, Kidney Research Institute, Medical Research Center, Seoul National University College of Medicine, 103 Daehak-ro, Jongno-gu, Seoul 110-799, Korea. Tel: +82-2-2072-1767, Fax: +82-2-743-5530, blue7270@snu.ac.kr
© 2012 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background
- Clear cell papillary renal cell carcinoma (CCPRCC) is a recently established subtype of renal epithelial tumor. The aim of this study was to identify the diagnostic criteria of CCPRCC with an emphasis on immunohistochemical studies, and to report three cases with concurrent other-type renal cell carcinoma (RCC).
-
Methods
- A total of 515 RCC patients that consecutively underwent surgical resection at Seoul National University Hospital from 1 January 2010 to 31 December 2011 were screened. Each case was reviewed based on the histologic features and was evaluated immunohistochemically.
-
Results
- A total of 15 CCPRCCs were identified, which composed 2.9% of the total RCCs. The mean age was 52 years, and the average tumor size was 1.65 cm. All 15 cases showed low nuclear grade, no lymph node metastasis and no distant metastasis. The CCPRCCs showed variable architectural patterns including cystic, trabecular, papillary, and acinar. All of the cases showed moderate to intense immunoreactivity for cytokeratin 7 (CK7). CD10 was negative or showed focal weak positivity. Three cases had concurrent other-type RCC, including a clear cell RCC and an acquired cystic disease-associated RCC.
-
Conclusions
- The strong CK7 and negative or focal weak CD10 expression will be useful for the diagnosis of CCPRCC.
- Screening and inclusion criteria
- A total of 515 patients with RCC who underwent radical or partial nephrectomy between 1 January 2010 and 31 December 2011 at Seoul National University Hospital were included in this study. Each RCC case was re-evaluated with regard to the RCC subtype; with reference to the "2009 update on the classification of renal epithelial tumors in adults."2 We retrieved the CCPRCC cases based on the pathologic features listed in several articles.9,12-14 RCCs with a primarily tubulopapillary architecture, clear cytoplasm, fibrous capsules, and characteristic nuclear alignment were considered the microscopic features of CCPRCCs. For a more accurate and precise retrieval, we performed CK7 and CD10 immunohistochemical staining on all 515 RCC cases. The cases with the appropriate histopathologic features, strong CK7 expression, and negative or focal weak CD10 expression were included in this study.
- Clinicopathologic evaluation
- A total of 15 cases of CCPRCC were identified. Tumor staging was defined according to the 2010 tumor-lymph nodes-metastasis (TNM) classification system,17 and nuclear grading was reviewed as in Fuhrman et al.18 Patient age, gender, and ESRD association were evaluated. RCC recurrence or metastasis was determined according to the clinical and radiographic findings. In each tumor section, we evaluated the histologic features that are common in CCPRCC. This study was approved by the Institutional Review Board of Seoul National University Hospital.
- Immunohistochemistry
- The immunohistochemical staining was performed on each representative slide of the 15 CCPRCCs. We evaluated immunoreactivity for CK7 (1:300, Dako, Glostrup, Denmark), CD10 (ready-to-use, Novocastra, Newcastle, UK), alpha-methylacyl-CoA racemase (AMACR; 1:300, Dako), TFE3 (1:1,500, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and vimentin (VT; 1:500, Dako). Each slide was dewaxed and rehydrated in a graded series of alcohol solutions. Immunohistochemical staining was performed using the Bond-Max autostainer (Leica Microsystems, Bannockburn, IL, USA) for CK7, CD10, TFE3, and VT, and the Dako Autostainer Link 48 (Dako Corp., Carpintera, CA, USA) was used for AMACR. The binding of the primary antibody was detected using the Bond polymer refine detection kit (Leica Microsystems) or the Dako EnVision Flex Kit according to the manufacturers' instructions.
MATERIALS AND METHODS
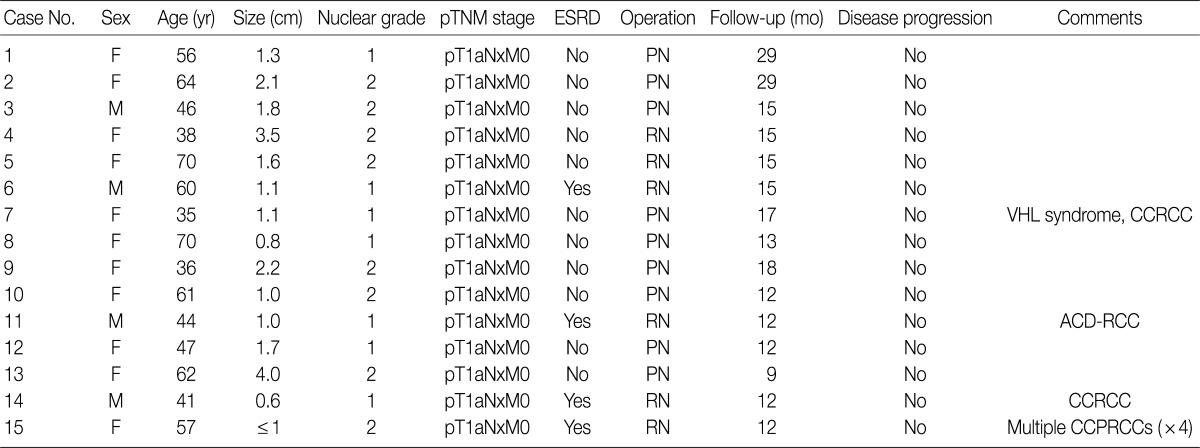
- The clinical and pathological characteristics of the CCPRCC patients
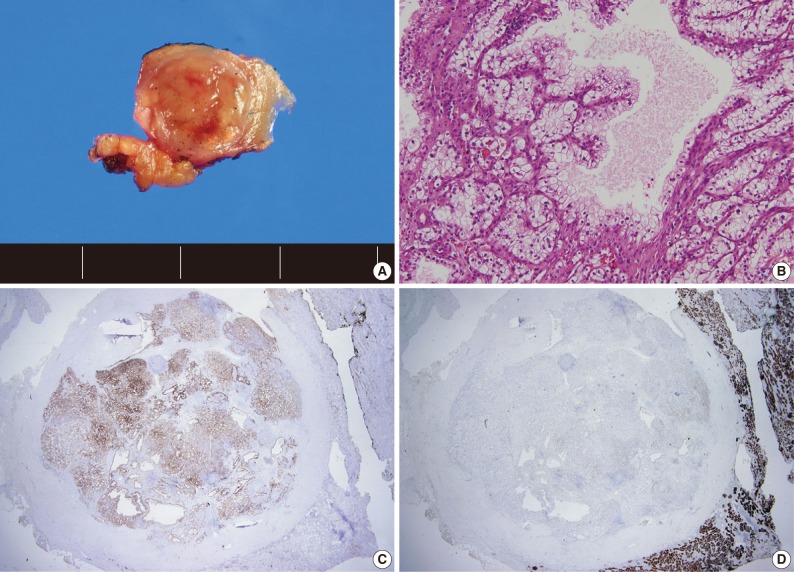
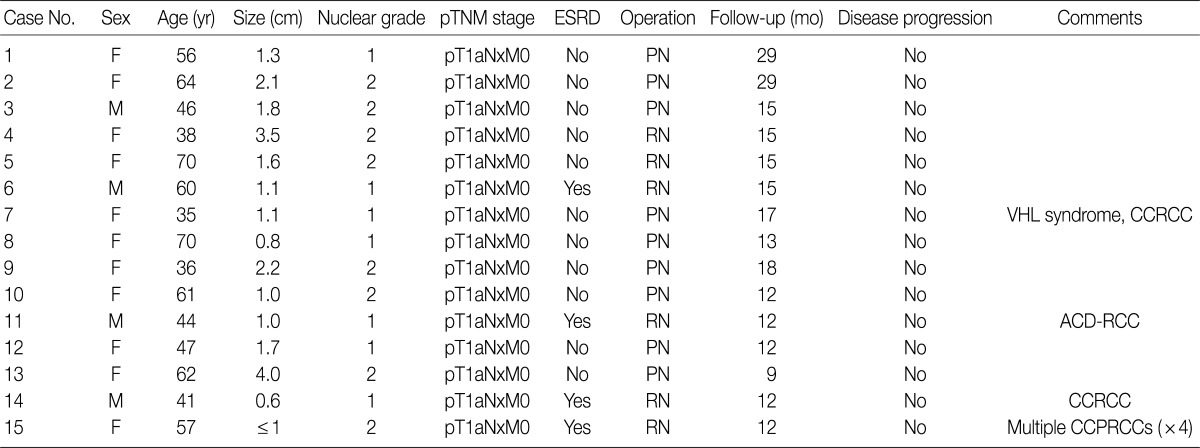
- A total of 15 patients who underwent surgical resection and were diagnosed with CCPRCC were analyzed in this study. The clinicopathological characteristics of the patients are summarized in Table 1. The patients included 4 men and 11 women. The mean age was 52 years (range, 35 to 70 years), and the mean tumor size was 1.65 cm (range, 0.6 to 4.0 cm). Lymph node metastasis and distant metastasis were not observed. All cases were pT1aNxM0, and thus stage I. Of the 15 cases, 8 (53.3%) showed Fuhrman nuclear grade 2, and the remaining 7 (46.7%) were Fuhrman nuclear grade 1. An association with ESRD was identified in 4 cases (26.7%). The mean follow-up duration was 15.6 months (range, 9 to 29 months), and there was no case with disease progression or metastasis. Lymph node dissection was not performed in any of the 15 cases, and was classified as pNx according to the 2010 TNM classification system.17 A representative gross photograph and microscopic findings are shown in Fig. 1.
- Multiple RCC cases
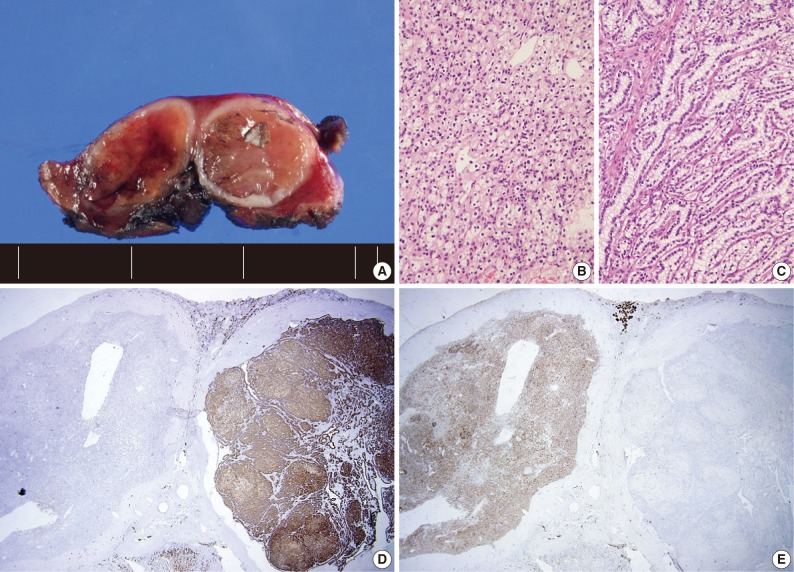
- Four patients had multiple RCCs. Among them, 3 patients had CCPRCC with other-type RCC. Two patients had CCPRCC and concurrent CCRCC. One patient was clinically diagnosed with VHL syndrome (Fig. 2), and had a family history of VHL syndrome; the patient's father and brother were also affected. The patient underwent surgery for spinal capillary hemangioblastoma and had a history of a brain tumor. The patient also had a pancreatic cyst. Another patient had CCPRCC and a concurrent acquired cystic disease-associated RCC (ACD-RCC). The remaining patient showed four CCPRCCs in the same kidney and also suffered from ESRD.
- Histopathologic features of the 15 CCPRCC cases
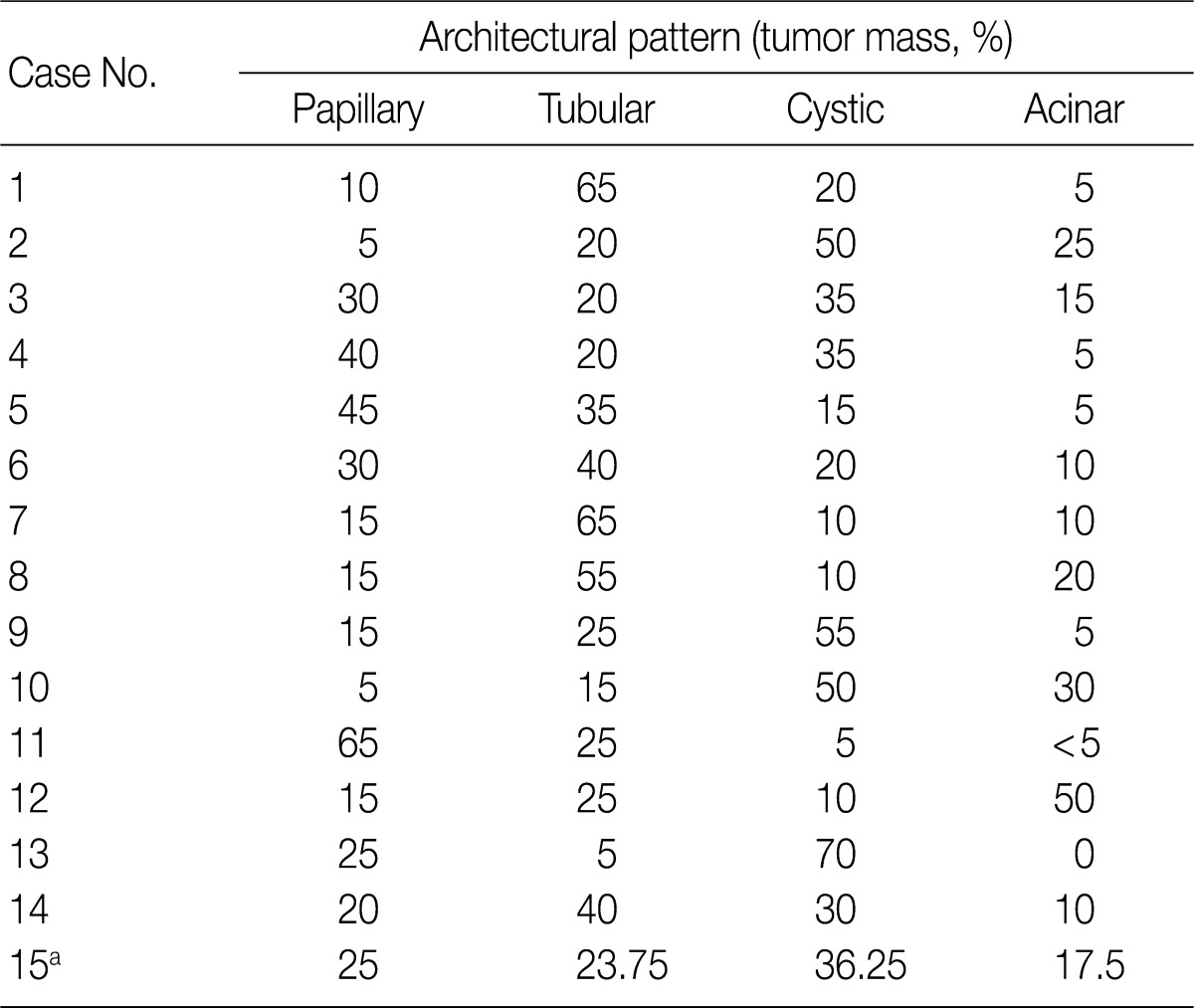
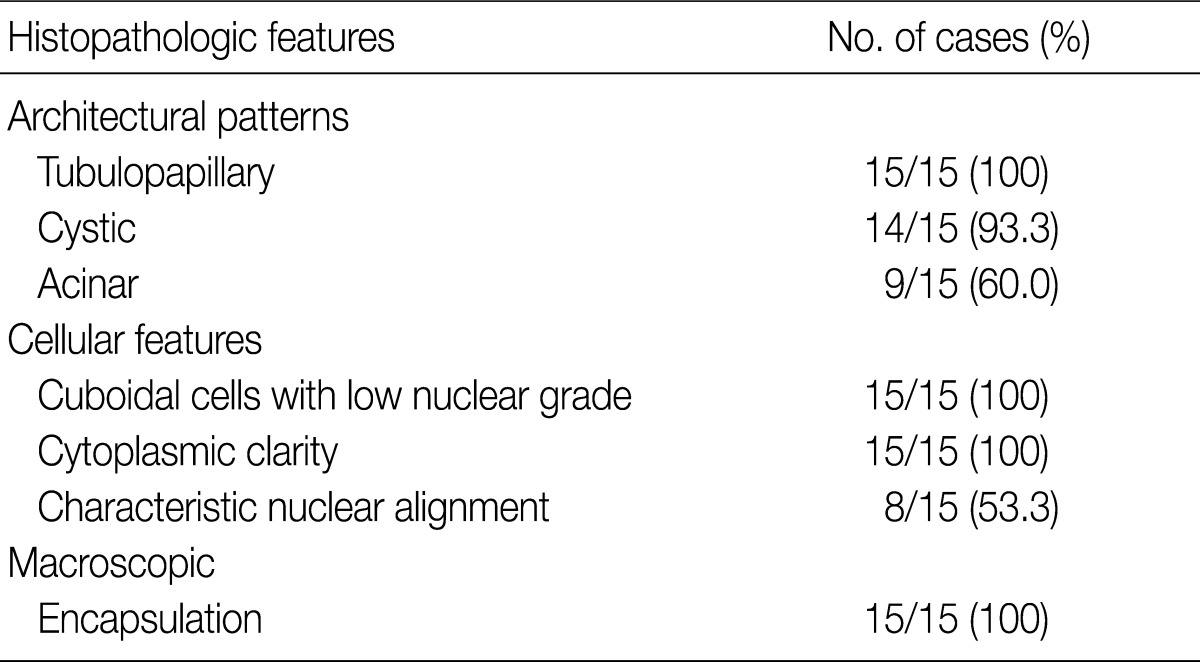
- The architectural patterns (papillary, tubular, cystic, and acinar) were examined and are summarized in Table 2. We also evaluated the cytoplasmic clarity, characteristic nuclear arrangement, and encapsulation. These results are summarized in Table 3. A tubulopapillary architecture was identified in all 15 cases (100%), a cystic architecture in 14 cases (93.3%), and an acinar architecture in 9 cases (60.0%). All of the tumor cells were cuboidal with a low Fuhrman nuclear grade and clear cytoplasm. The characteristic nuclear alignment away from the basal aspect was identified in 8 cases (53.3%). All of the tumors were encapsulated by a fibrous capsule.
- Immunohistochemical features of the 15 CCPRCC cases
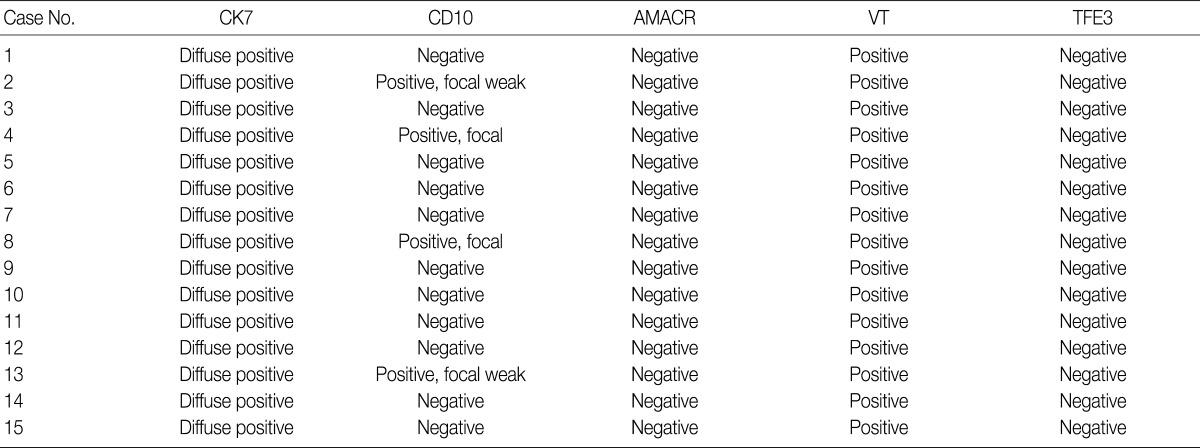
- We assessed the immunohistochemical profile of CK7, CD10, AMACR, VT, and TFE3 in the 15 CCPRCC cases (Table 4). All 15 cases (100%) showed strong immunoreactivity for CK7. Eleven cases (73.3%) were negative for CD10, and 4 cases (26.7%) showed focal positivity. All of the cases were negative for AMACR and TFE3. All 15 cases were positive for VT immunoreactivity.
RESULTS
- Due to the development of imaging modalities and the increased number of cases of early detection of RCC, the relative proportion of small RCCs has increased. Without an understanding of CCPRCC and its helpful diagnostic features, RCC can be misdiagnosed as CCRCC. Histopathologically, CCPRCC has shown a tubular and papillary architecture, tumor cells with clear cytoplasms, and a low Fuhrman nuclear grade; the characteristic nuclear arrangement away from the basement membrane has also been observed.9,11,15
- CCPRCC was initially thought to occur in association with ESRD,7 and was regarded as a renal cell neoplasm in ESRD.2 However, several studies have reported that CCPRCC cases can occur in patients without ESRD.9,10,12,13 In our study, ESRD was associated with CCPRCC in only 4 cases (26.7%).
- Most small CCRCCs show a low Fuhrman nuclear grade and display variable architectural patterns including solid, acinar, cystic, tubular, and papillary patterns.1,2 CCPRCCs can also show variable architectural patterns including tubulopapillary, solid, acinar, and cystic patterns.7-15 Moreover, the so-called characteristic nuclear arrangement of CCPRCC has not been identified in all cases and cannot be used to verify a diagnosis of CCPRCC. For instance, in our study, the characteristic nuclear alignment was identified in only 8 cases (53.3%). Therefore, a differential diagnosis between CCRCC and CCPRCC can be very difficult.
- Renal carcinomas associated with Xp11.2 translocations are less commonly encountered in a differential diagnosis of CCPRCC.19 Typically, this entity presents at an advanced stage, and it is rarely detected in an early stage. Renal carcinomas associated with Xp11.2 translocations can show a papillary architecture and clear cells,1,19 and immunohistochemical staining for TFE3 and translocation studies can lead to a correct diagnosis. Papillary adenoma of the kidney would also be considered a differential diagnosis of CCPRCC. Papillary adenoma shows a papillary or tubular architecture and has a low nuclear grade. By definition, the diameter of a papillary adenoma is less than 5 mm. Though papillary adenoma has a similar architecture to CCPRCC and shows CK7 positivity, the tumor cells have features more similar to PRCC than CCPRCC, and the cytoplasm is not clear. These cytologic features may be helpful for the differential diagnosis of CCPRCC.
- In our study, we identified the usefulness of immunohistochemical staining in the diagnosis of CCPRCC. All 15 CCPRCC cases showed diffuse positivity for CK7 and negative or focal weak positivity for CD10. In general, CCRCCs demonstrate strong and diffuse staining for CD10 and negative or focal weak staining for CK7,1,20 while CCPRCCs show the opposite results.9,10,14,15 Some histopathologic features, including a well-encapsulated mass, a primarily tubulopapillary architecture, and characteristic nuclear alignment, may increase the possibility of diagnosing CCPRCC, but these findings are somewhat subjective and do not always exist. However, CCRCCs can also show those histopathologic features. Therefore, a histologic examination may not be enough for the differential diagnosis between CCPRCC and CCRCC, and additional immunohistochemical staining for CK7 and CD10 may be very useful.
- As far as we know, there have not been any reports of CCPRCC in Korea. Furthermore, there have been no reports on the proportion of CCPRCCs among all RCCs worldwide. In our study, we evaluated cases of surgically resected RCCs over a two-year period. The proportion of CCPRCCs among all of the RCCs was 2.9%. Compared with the frequency of chromophobe RCC, which accounts for approximately 5% of all RCCs,1 CCPRCC was not rare. Without an understanding of CCPRCC and without precise diagnostic histologic and immunohistochemical criteria, CCPRCCs can be misdiagnosed as CCRCCs.
- We also assessed the prognosis of 15 CCPRCC cases. Though the mean follow-up duration was short (15.6 months), all 15 CCPRCC cases showed a favorable prognosis with no disease progression or no metastatis. These results were consistent with previous articles that have reported no recurrence or metastases in CCPRCC patients.7,9,12 There was one article that evaluated the prognosis of CCRCC according to pT staging.21 Though the article was based on the 5th American Joint Committee on Cancer (AJCC) cancer staging manual, and pT1 included tumors less than 7 cm; the 1 year cancer-specific survival rate of pT1 CCRCC was about 95% and 5 year survival rate was 88.7%.
- Several reports have shown multiple CCPRCCs in the same patients. Gobbo et al.9 presented one patient with three CCPRCCs and ESRD. Aydin et al.12 included three patients with bilateral CCPRCC, and each patient had two CCPRCCs; the study also included one VHL patient with one CCPRCC. The study by Adam et al.13 contained four patients with multiple CCPRCCs; two patients had two bilateral CCPRCCs and impaired renal functions, and one patient had eight unilateral CCPRCCs and normal renal function. The fourth patient had multiple bilateral CCPRCCs and suffered from ESRD. Kuroda et al.16 reported three concurrent RCCs in a patient with impaired renal function. Among them, two tumors were CCRCCs, and one tumor was a CCPRCC.
- In our cases, there were four multiple RCC cases. Interestingly, there were three cases that occurred with concurrent other-type RCC. One patient was clinically diagnosed with VHL syndrome with a family history of VHL syndrome (the patient's father and brother), capillary hemangioblastoma of the spine, and a brain tumor. As far as we know, this is the second CCPRCC case that has been reported to occur in a VHL syndrome patient. This patient showed normal renal function and had two RCCs of different histologic types, CCPRCC and CCRCC. A genetic study of the VHL gene mutation may show interesting results. However, we did not perform a genetic study of the VHL gene mutation in the CCRCC and CCPRCC cases in this study. We believe that this is a limitation of our study. Because the molecular pathogenesis of CCRCC and CCPRCC is considered to be different, investigating the VHL gene mutation status in both types of RCC may be interesting.
- The second patient showed impaired renal function, and had ACD-RCC and CCPRCC. The third patient also showed impaired renal function, and had CCRCC and CCPRCC.
- At the molecular level, CCPRCC showed different genetic alterations from CCRCC and PRCC.12-14,16 The loss of chromosome 3p, where the VHL gene and polybromo-1 (PBRM1) gene are located, is associated with CCRCC.1,22 Another genetic change identified in CCRCC is the loss of 9p and 14q.2 In PRCC cases, trisomy or tetrasomy of chromosome 7, trisomy 17, and a loss of the Y chromosome are characteristic features.23,24 However, CCPRCC does not have those genetic alterations.12-14,16 In CCPRCC, chromosomal alterations have been reported in a few cases. Aydin et al.12 reported low copy number gains at chromosomes 7 and 17 in one case of CCPRCC among 36 cases. Additionally, Kuroda et al.16 found polysomy for chromosome 7 and monosomy for chromosomes 17, 16, and 20 in one CCPRCC case. These results showed that CCPRCC is an entity unique from CCRCC and PRCC.
- In summary, CCPRCC is a recently established type of renal epithelial tumor and one of the primary differential diagnoses of small RCCs with clear cytoplasms. The observation of strong CK7 expression and a negative or weak CD10 expression pattern is very useful for the differential diagnosis between CCRCC and CCPRCC.
DISCUSSION
Acknowledgments
Acknowledgments
- 1. Eble JN, Sauter G, Epstein JI, Sesterhenn IA. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and mela genital organs. 2004; Lyon: IARC Press.
- 2. Lopez-Beltran A, Carrasco JC, Cheng L, Scarpelli M, Kirkali Z, Montironi R. 2009 update on the classification of renal epithelial tumors in adults. Int J Urol 2009; 16: 432-443. ArticlePubMed
- 3. Srigley JR, Delahunt B. Uncommon and recently described renal carcinomas. Mod Pathol 2009; 22(Suppl 2): S2-S23. ArticlePubMedPDF
- 4. Amin MB, Tamboli P, Javidan J, et al. Prognostic impact of histologic subtyping of adult renal epithelial neoplasms: an experience of 405 cases. Am J Surg Pathol 2002; 26: 281-291. PubMed
- 5. Ficarra V, Brunelli M, Cheng L, et al. Prognostic and therapeutic impact of the histopathologic definition of parenchymal epithelial renal tumors. Eur Urol 2010; 58: 655-668. ArticlePubMed
- 6. Capitanio U, Cloutier V, Zini L, et al. A critical assessment of the prognostic value of clear cell, papillary and chromophobe histological subtypes in renal cell carcinoma: a population-based study. BJU Int 2009; 103: 1496-1500. ArticlePubMed
- 7. Tickoo SK, dePeralta-Venturina MN, Harik LR, et al. Spectrum of epithelial neoplasms in end-stage renal disease: an experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol 2006; 30: 141-153. PubMed
- 8. Michal M, Hes O, Havlicek F. Benign renal angiomyoadenomatous tumor: a previously unreported renal tumor. Ann Diagn Pathol 2000; 4: 311-315. ArticlePubMed
- 9. Gobbo S, Eble JN, Grignon DJ, et al. Clear cell papillary renal cell carcinoma: a distinct histopathologic and molecular genetic entity. Am J Surg Pathol 2008; 32: 1239-1245. PubMed
- 10. Mai KT, Kohler DM, Belanger EC, Robertson SJ, Wang D. Sporadic clear cell renal cell carcinoma with diffuse cytokeratin 7 immunoreactivity. Pathology 2008; 40: 481-486. ArticlePubMed
- 11. Michal M, Hes O, Nemcova J, et al. Renal angiomyoadenomatous tumor: morphologic, immunohistochemical, and molecular genetic study of a distinct entity. Virchows Arch 2009; 454: 89-99. ArticlePubMedPDF
- 12. Aydin H, Chen L, Cheng L, et al. Clear cell tubulopapillary renal cell carcinoma: a study of 36 distinctive low-grade epithelial tumors of the kidney. Am J Surg Pathol 2010; 34: 1608-1621. ArticlePubMed
- 13. Adam J, Couturier J, Molinié V, Vieillefond A, Sibony M. Clear-cell papillary renal cell carcinoma: 24 cases of a distinct low-grade renal tumour and a comparative genomic hybridization array study of seven cases. Histopathology 2011; 58: 1064-1071. ArticlePubMed
- 14. Rohan SM, Xiao Y, Liang Y, et al. Clear-cell papillary renal cell carcinoma: molecular and immunohistochemical analysis with emphasis on the von Hippel-Lindau gene and hypoxia-inducible factor pathway-related proteins. Mod Pathol 2011; 24: 1207-1220. ArticlePubMedPDF
- 15. Gobbo S, Eble JN, Maclennan GT, et al. Renal cell carcinomas with papillary architecture and clear cell components: the utility of immunohistochemical and cytogenetical analyses in differential diagnosis. Am J Surg Pathol 2008; 32: 1780-1786. PubMed
- 16. Kuroda N, Shiotsu T, Kawada C, et al. Clear cell papillary renal cell carcinoma and clear cell renal cell carcinoma arising in acquired cystic disease of the kidney: an immunohistochemical and genetic study. Ann Diagn Pathol 2011; 15: 282-285. ArticlePubMed
- 17. Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A. AJCC cancer staging manual. 2009; 7th ed. New York: Springer.
- 18. Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol 1982; 6: 655-663. ArticlePubMed
- 19. Ross H, Martignoni G, Argani P. Renal cell carcinoma with clear cell and papillary features. Arch Pathol Lab Med 2012; 136: 391-399. ArticlePubMedPDF
- 20. Zhou M, Roma A, Magi-Galluzzi C. The usefulness of immunohistochemical markers in the differential diagnosis of renal neoplasms. Clin Lab Med 2005; 25: 247-257. ArticlePubMed
- 21. Cheville JC, Lohse CM, Zincke H, Weaver AL, Blute ML. Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol 2003; 27: 612-624. ArticlePubMed
- 22. Varela I, Tarpey P, Raine K, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011; 469: 539-542. PubMedPMC
- 23. Kovacs G, Fuzesi L, Emanual A, Kung HF. Cytogenetics of papillary renal cell tumors. Genes Chromosomes Cancer 1991; 3: 249-255. ArticlePubMed
- 24. Tan MH, Rogers CG, Cooper JT, et al. Gene expression profiling of renal cell carcinoma. Clin Cancer Res 2004; 10(18 Pt 2): 6315S-6321S. ArticlePDF
REFERENCES
CCPRCC, clear cell papillary renal cell carcinoma; TNM, tumor-lymph nodes-metastasis; ESRD, end-stage renal disease; F, female; PN, partial nephrectomy; M, male; RN, radical nephrectomy; VHL, von Hippel-Lindau syndrome; CCRCC, clear cell renal cell carcinoma; ACD-RCC, acquired cystic disease-associated RCC.
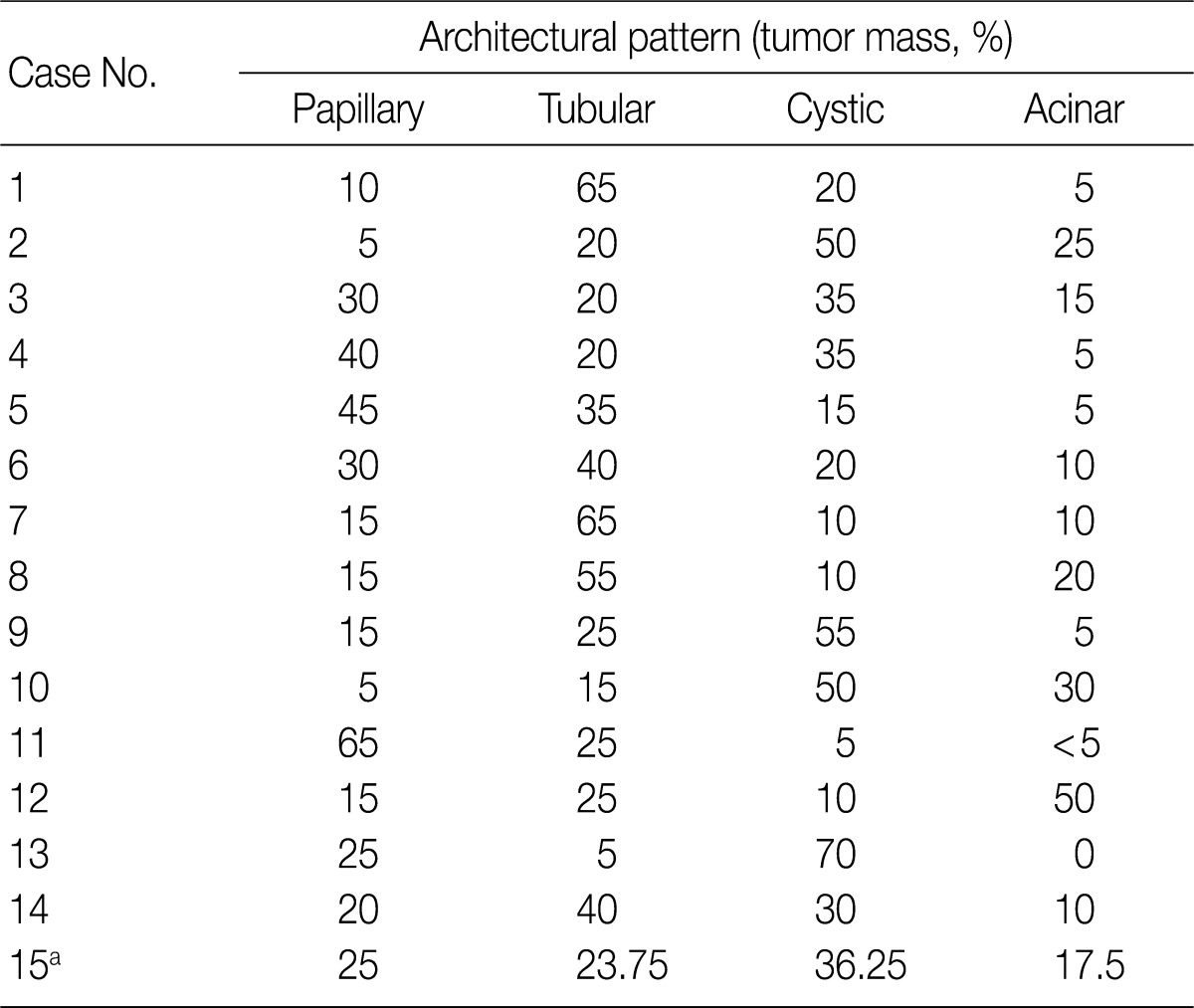
Figure & Data
References
Citations
- Vascular, adipose tissue, and/or calyceal invasion in clear cell tubulopapillary renal cell tumour: potentially problematic diagnostic scenarios
Ankur R Sangoi, Harrison Tsai, Lara Harik, Jonathan Mahlow, Maria Tretiakova, Sean R Williamson, Michelle S Hirsch
Histopathology.2024; 84(7): 1167. CrossRef - Clinical features and Surgical Outcome of Clear Cell Papillary Renal Cell Tumor: result from a prospective cohort
Si Hyun Kim, Jang Hee Han, Seung-hwan Jeong, Hyeong Dong Yuk, Ja Hyeon Ku, Cheol Kwak, Hyeon Hoe Kim, Kyung Chul Moon, Chang Wook Jeong
BMC Urology.2023;[Epub] CrossRef - Coexistence of multiple clear cell papillary renal cell carcinoma with renal oncocytoma: a case report
Amine Hermi, Ahmed Saadi, Seif Mokadem, Ahlem Blel, Marouene Chakroun, Mohamed Riadh Ben Slama
Annals of Medicine & Surgery.2023; 85(5): 2017. CrossRef - Renal Cell Carcinoma in End-Stage Renal Disease: A Review and Update
Ziad M. El-Zaatari, Luan D. Truong
Biomedicines.2022; 10(3): 657. CrossRef - The Clinicopathologic and Molecular Landscape of Clear Cell Papillary Renal Cell Carcinoma: Implications in Diagnosis and Management
Stanley Weng, Renzo G. DiNatale, Andrew Silagy, Roy Mano, Kyrollis Attalla, Mahyar Kashani, Kate Weiss, Nicole E. Benfante, Andrew G. Winer, Jonathan A. Coleman, Victor E. Reuter, Paul Russo, Ed Reznik, Satish K. Tickoo, A. Ari Hakimi
European Urology.2021; 79(4): 468. CrossRef - Clear cell papillary renal cell carcinoma: Characteristics and survival outcomes from a large single institutional series
James E. Steward, Sean Q. Kern, Liang Cheng, Ronald S. Boris, Yan Tong, Clint D. Bahler, Timothy A. Masterson, K. Clint Cary, Hristos Kaimakliotis, Thomas Gardner, Chandru P. Sundaram
Urologic Oncology: Seminars and Original Investigations.2021; 39(6): 370.e21. CrossRef - Clear cell papillary renal cell carcinoma: an update after 15 years
Sean R. Williamson
Pathology.2021; 53(1): 109. CrossRef - Clear Cell Papillary Renal Cell Carcinoma
Jianping Zhao, Eduardo Eyzaguirre
Archives of Pathology & Laboratory Medicine.2019; 143(9): 1154. CrossRef - Clear cell papillary renal cell carcinoma – An indolent subtype of renal tumor
Wei-Jen Chen, Chin-Chen Pan, Shu-Huei Shen, Hsiao-Jen Chung, Chih-Chieh Lin, Alex T.L. Lin, Yen-Hwa Chang
Journal of the Chinese Medical Association.2018; 81(10): 878. CrossRef - Clear cell papillary renal cell carcinoma: A case report and review of the literature
Sung Han Kim, Whi-An Kwon, Jae Young Joung, Ho Kyung Seo, Kang Hyun Lee, Jinsoo Chung
World Journal of Nephrology.2018; 7(8): 155. CrossRef - Clinical features and survival analysis of clear cell papillary renal cell carcinoma: A 10‑year retrospective study from two institutions
Yiqiu Wang, Ying Ding, Jian Wang, Min Gu, Zengjun Wang, Chao Qin, Conghui Han, Hongxia Li, Xia Liu, Pengfei Wu, Guangchao Li
Oncology Letters.2018;[Epub] CrossRef - A contemporary series of renal masses with emphasis on recently recognized entities and tumors of low malignant potential: A report based on 624 consecutive tumors from a single tertiary center
Maria Rosaria Raspollini, Ilaria Montagnani, Rodolfo Montironi, Liang Cheng, Guido Martignoni, Andrea Minervini, Sergio Serni, Giulio Nicita, Marco Carini, Antonio Lopez-Beltran
Pathology - Research and Practice.2017; 213(7): 804. CrossRef - Renal Neoplasms With Overlapping Features of Clear Cell Renal Cell Carcinoma and Clear Cell Papillary Renal Cell Carcinoma
Hari P. Dhakal, Jesse K. McKenney, Li Yan Khor, Jordan P. Reynolds, Cristina Magi-Galluzzi, Christopher G. Przybycin
American Journal of Surgical Pathology.2016; 40(2): 141. CrossRef - New and emerging renal tumour entities
Naoto Kuroda, Ondřej Hess, Ming Zhou
Diagnostic Histopathology.2016; 22(2): 47. CrossRef - Immunohistochemical Panel for Differentiating Renal Cell Carcinoma with Clear and Papillary Features
Hanan AlSaeid Alshenawy
Pathology & Oncology Research.2015; 21(4): 893. CrossRef - Immunohistochemical panel for differentiating renal cell carcinoma with clear and papillary features
Hanan AlSaeid Alshenawy
Journal of Microscopy and Ultrastructure.2015; 3(2): 68. CrossRef - Clear Cell-Papillary Renal Cell Carcinoma of the Kidney Not Associated With End-stage Renal Disease
Manju Aron, Elena Chang, Loren Herrera, Ondrej Hes, Michelle S. Hirsch, Eva Comperat, Philippe Camparo, Priya Rao, Maria Picken, Michal Michal, Rodolfo Montironi, Pheroze Tamboli, Federico Monzon, Mahul B. Amin
American Journal of Surgical Pathology.2015; 39(7): 873. CrossRef - Papillary or pseudopapillary tumors of the kidney
Fang-Ming Deng, Max X. Kong, Ming Zhou
Seminars in Diagnostic Pathology.2015; 32(2): 124. CrossRef - Do Clear Cell Papillary Renal Cell Carcinomas Have Malignant Potential?
Mairo L. Diolombi, Liang Cheng, Pedram Argani, Jonathan I. Epstein
American Journal of Surgical Pathology.2015; 39(12): 1621. CrossRef - Targeted next‐generation sequencing and non‐coding RNA expression analysis of clear cell papillary renal cell carcinoma suggests distinct pathological mechanisms from other renal tumour subtypes
Charles H Lawrie, Erika Larrea, Gorka Larrinaga, Ibai Goicoechea, María Arestin, Marta Fernandez‐Mercado, Ondrej Hes, Francisco Cáceres, Lorea Manterola, José I López
The Journal of Pathology.2014; 232(1): 32. CrossRef - Clear cell papillary renal cell carcinoma is the fourth most common histologic type of renal cell carcinoma in 290 consecutive nephrectomies for renal cell carcinoma
Haijun Zhou, Shaojiang Zheng, Luan D. Truong, Jae Y. Ro, Alberto G. Ayala, Steven S. Shen
Human Pathology.2014; 45(1): 59. CrossRef - Clear cell papillary renal cell carcinoma: Incidence, morphological features, immunohistochemical profile, and biologic behavior: A single institution study
Borislav A. Alexiev, Cinthia B. Drachenberg
Pathology - Research and Practice.2014; 210(4): 234. CrossRef - MRI Phenotype in Renal Cancer
Naomi Campbell, Andrew B. Rosenkrantz, Ivan Pedrosa
Topics in Magnetic Resonance Imaging.2014; 23(2): 95. CrossRef
 PubReader
PubReader Cite this Article
Cite this Article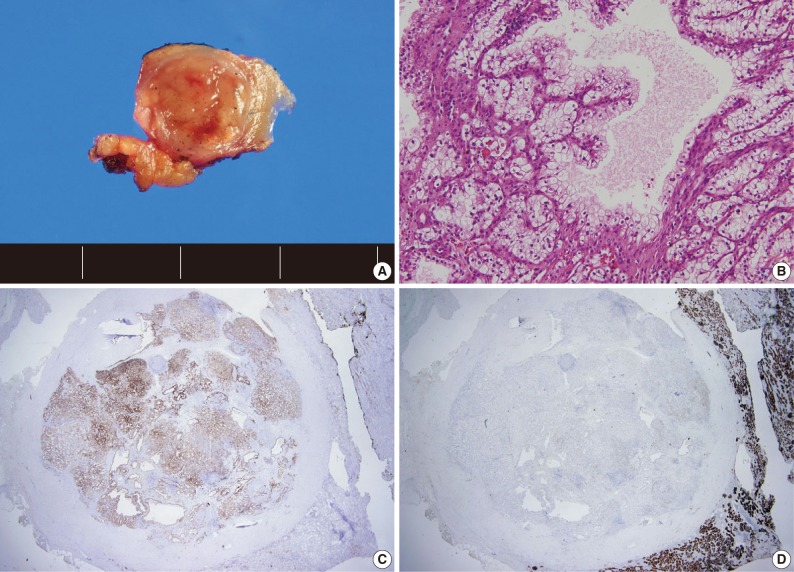
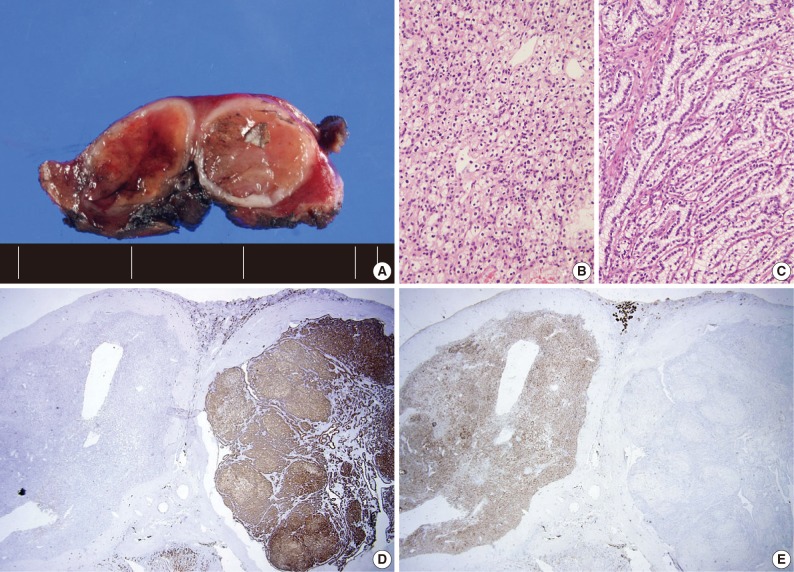


Fig. 1
Fig. 2
CCPRCC, clear cell papillary renal cell carcinoma; TNM, tumor-lymph nodes-metastasis; ESRD, end-stage renal disease; F, female; PN, partial nephrectomy; M, male; RN, radical nephrectomy; VHL, von Hippel-Lindau syndrome; CCRCC, clear cell renal cell carcinoma; ACD-RCC, acquired cystic disease-associated RCC.
CCPRCCs, clear cell papillary renal cell carcinomas. aCase No. 15 contains four CCPRCCs and the architectural proportion is calculated the average.
CCPRCCs, clear cell papillary renal cell carcinomas. aThe significant percent of proportion of each category is considered more than 10%.
CCPRCC, clear cell papillary renal cell carcinoma; AMACR, alpha-methylacyl-CoA racemase; VT, vimentin; TFE3, transcription factor E3.

