EBV-Positive T/NK-Cell Lymphoproliferative Disease of Childhood
Article information
Abstract
Background
Epstein-Barr virus (EBV)-associated hemophagocytic lymphohistiocytosis (HLH), EBV-positive systemic T-cell lymphoproliferative disease (STLPD) of childhood, and chronic active EBV (CAEBV) infection may develop after primary EBV infection. This study reviewed the clinicopathological spectrum of EBV-associated T- and natural killer (NK)-cell LPD, including STLPD and CAEBV infection, with an analysis of T-cell clonality.
Methods
Clinicopathological features of seven patients with EBV-associated HLH or STLPD and 12 patients with CAEBV infection were reviewed. Immunohistochemical staining and a T-cell receptor (TCR) gene rearrangement study were performed.
Results
STLPD and EBV-positive HLH showed significantly overlapping clinicopathological findings. One patient with STLPD and one patient with EBV-positive HLH demonstrated moderate to severe atypia of the infiltrating lymphocytes, whereas the remaining patients lacked significant atypia. Twelve patients had CAEBV infection, four of whom suffered mosquito-bite hypersensitivity, five showed NK lymphocytosis, and one suffered hydroa vacciniforme. Infiltrating lymphocytes were predominantly small and devoid of atypia. Hemophagocytic histiocytosis was found in seven of 11 patients. Monoclonality was detected in three (50%) of the six patients with successful TCR gene analysis.
Conclusions
EBV-positive HLH and STLPD share similar clinicopathological findings and may constitute a continuous spectrum of acute EBV-associated T- or NK-cell proliferative disorders. The distinction of EBV-positive T-cell LPD from EBV-positive HLH may be difficult during routine diagnoses because of the technical limitations of clonality assessment.
Epstein-Barr virus (EBV) is a ubiquitous herpes virus, and over 95% of the human population is infected during their lifetime. Primary EBV infection is usually asymptomatic, and those infected recover completely. However, some people develop infectious mononucleosis, which is characterized by a triad of symptoms including fever, pharyngitis, and lymphadenopathy which last for one to four weeks. Infectious mononucleosis is usually resolved within one to two months without treatment.1
Uncommonly, primary EBV infection is complicated by hemophagocytic syndrome, which has been called EBV-associated hemophagocytic lymphohistiocytosis (HLH). EBV-associated HLH is defined by the criteria for HLH-94 and HLH-2004,2,3 and patients must fulfill five of eight criteria, including fever, splenomegaly, bicytopenia, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis, low/absent natural killer (NK)-cell activity, hyperferritinemia, and high soluble interleukin-2 receptor levels, unless the family history or molecular diagnosis is consistent with HLH. By definition, the patient has no evidence of malignancy, but some patients may transform to T- or NK-cell malignancy during the course of the disease.2,3
EBV-positive systemic T-cell lymphoproliferative disease (LPD) of childhood may develop after primary EBV infection and has been reported previously under the names of fulminant EBV-positive T-cell LPD of childhood, sporadic fatal infectious mononucleosis, fulminant hemophagocytic syndrome, fatal EBV-associated hemophagocytic syndrome, and severe chronic active EBV (CAEBV) infection.4,5 The disease shares significant clinicopathological similarities with EBV-associated HLH, but is defined as a clonal disease of EBV-infected T-cells with an activated cytotoxic phenotype in the 2008 World Health Organization (WHO) classification.4 The disease occurs most often in children and young adults after a primary EBV infection, but can also occur in adult patients.5 Histologically, the proliferating cells often lack cytologic atypia. Hemophagocytic histiocytosis in the hepatic sinusoids or bone marrow is the main histological change. Even the infiltrating cells appear benign, and the patients follow a fulminant clinical course arising from hemophagocytic syndrome and multiorgan failure.4
A minority of patients with primary EBV infection develop CAEBV infection, which is initially defined by chronic or recurrent infectious mononucleosis-like symptoms that persist for at least six months and by an unusual pattern of anti-EBV antibodies.1 CAEBV infection is not a simple EBV infection, but a LPD induced by EBV infection. Patients with CAEBV infection may present with a variety of clinical signs and symptoms, including fever, hepatosplenomegaly, lymphadenopathy, and skin lesions, as well as hypersensitivity to mosquito bites and hydroa vacciniforme.4 Laboratory findings include non-specific abnormalities, such as liver dysfunction, thrombocytopenia, anemia, and EBV-related abnormalities, including elevated antibody titers against viral capsid antigen and/or early antigen and an elevated EBV DNA load. The pathological changes are variable, ranging from reactive hyperplasia to monomorphic T- or NK-cell proliferation, which can be polyclonal or oligoclonal. Monoclonal T-cell LPD arising in CAEBV infection was classified as systemic T-cell lymphoproliferative disease (STLPD) of childhood in the 2008 WHO classification. The prognosis is variable, and a substantial proportion of patients die from the disease. The main causes of death are hemophagocytic syndrome and T- or NK-cell lymphoma. Adverse prognostic factors in CAEBV infection include the T-cell lineage, an age of onset older than eight years, and liver dysfunction.6
Because EBV-positive T- or NK-cell LPD of childhood shows variable histological changes and the infiltrating cells look deceptively benign, the distinction between reactive lymphoproliferation and aggressive neoplastic lymphoid proliferation is very difficult when based on histological changes. Clonality is regarded as a criterion distinguishing EBV-positive STLPD of childhood from other EBV-positive hemophagocytic syndromes and CAEBV infection,4 but the prognostic impact of T-cell or EBV clonality on EBV-associated LPD is unclear. The purpose of this study was to review the clinicopathological spectrum of EBV-associated T- and NK-cell LPD of childhood, including STLPD of childhood and CAEBV infection, with an analysis of T-cell clonality.
MATERIALS AND METHODS
Patients
CAEBV infection was defined as 1) persistent or recurrent symptoms related to EBV infection for more than three months; 2) high EBV genome levels in affected tissues or peripheral blood; 3) chronic illness that could not be explained by other known disease processes at diagnosis, and 4) no specific underlying immunological abnormality.7
To identify EBV-positive systemic T-cell LPD of childhood based on the 2008 WHO classification, all patients with EBV-positive hemophagocytic syndrome presenting at our institution were enrolled in the present study. Patient inclusion was based on the fulfillment of the diagnostic criteria for HLH-2004 in addition to the presence of a high viral load in the tissues or peripheral blood.3
The surgical pathology database of Samsung Medical Center from 1994 to 2012 was screened with keywords including "CAEBV," "EBV," and "hemophagocytosis." Of the 96 cases retrieved using "EBV" and "hemophagocytosis" as keywords, 84 patients were excluded because LPD had arisen in immunocompromised patients, such as those receiving organ transplantation or patients with congenital immune deficiency syndrome. Fifteen patients were diagnosed with CAEBV infection, and 12 patients had paraffin blocks available for examination. Among the remaining 12 patients, seven had paraffin blocks available for examination and were enrolled in the present study. Formalin-fixed paraffin-embedded blocks of liver, bone marrow, and lymph node tissues were used for further investigation.
Immunohistochemistry
Immunohistochemical analysis was performed on the paraffin sections using monoclonal and polyclonal antibodies to detect lineage-specific or lineage-characteristic antigens, including CD3 (Dakopatts, Copenhagen, Denmark), CD20 (Novocastra, Newcastle upon Tyne, UK), CD56 (Novocastra), CD4 (Novocastra), and CD8 (Novocastra).
T-cell receptor (TCR) gene rearrangement
For the polymerase chain reaction (PCR) amplification of the TCR γ locus, DNA was extracted from the paraffin-embedded formalin-fixed tissues with the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). Conventional PCR followed by single-stranded conformational polymorphism analysis was performed, as previously described.8 When conventional PCR for TCR γ genes produced negative results, further analysis was performed using the TCR β and γ primers, according to the recommendations of the BIOMED-2 protocols (Invivoscribe, San Diego, CA, USA).9
RESULTS
All patients (12 males and eight females) were Korean, ranging from 4 to 61 years of age.
EBV-positive hemophagocytic syndrome
TCR gene rearrangement
TCR gene rearrangement analysis revealed monoclonality in two patients (28.6%) and polyclonality in four patients (57.1%) (Fig. 1). The analysis of one patient failed because the DNA quality was poor. Two patients with T-cell monoclonality were classified as suffering systemic T-cell LPD and the remaining patients as suffering EBV-positive HLH.
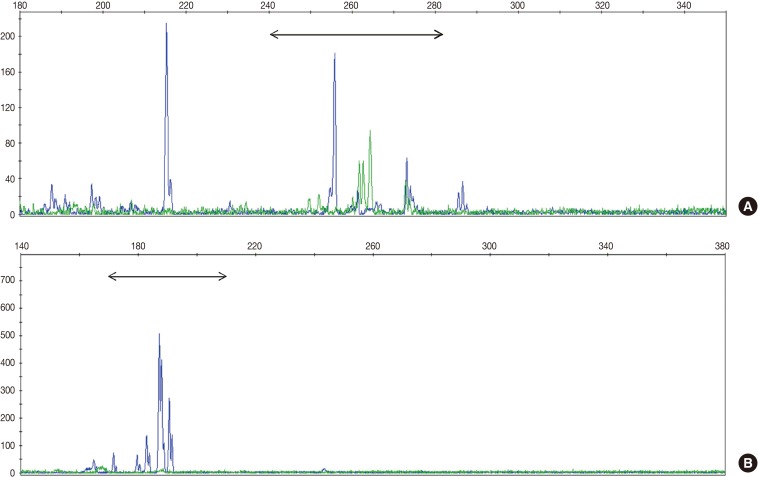
Clinicopathological findings of systemic T-cell LPD and EBV-positive HLH
Of the two patients with systemic T-cell LPD, one was a young female and the other an adult male. The EBV-positive HLH patients included two children, two young adults, and one elderly adult, with ages ranging from 4 to 61 years (median, 20 years). The clinicopathological findings for the two conditions were similar, and almost all patients had acute-onset fever. Their symptoms included lymph node enlargement and hepatosplenomegaly. The duration of the symptoms was two months in patients with systemic T-cell LPD and ranged from three days to one month (median, 1 month) in EBV-positive HLH patients. Two patients with systemic T-cell LPD received chemotherapy but died after two and three months, respectively. Among the five patients with EBV-positive HLH, four received chemotherapy or steroid and died after a median period of one month, whereas the one patient treated with the HLH-2004 regimen recovered completely and was alive at the final follow-up. The EBV load in the peripheral blood was analyzed using whole blood, and the loads during the clinical course are shown in Table 1. The levels were variable, and two patients with EBV-positive HLH showed high viral loads in their peripheral blood.

Clinicopathological findings of systemic T-cell lymphoproliferative disease and EBV-positive hemophagocytic lymphohistiocytosis
Biopsies were obtained from the bone marrow of six patients, lymph nodes of two patients, liver of one patient, and spleen of one patient. In the bone marrow, liver, and spleen samples, the number of lymphocytes was slightly increased and showed mild to equivocal atypia. Hemophagocytic histiocytosis was observed in all patients. The lymph node biopsies from patient 1 (systemic T-cell LPD) and patient 3 (EBV-positive HLH) demonstrated diffuse effacement of the nodal architecture by the infiltration of monotonous medium-sized atypical lymphocytes (Fig. 2). The lymphocytes had hyperchromatic nuclei and irregular nuclear contours. At the first liver biopsy of patient 3, EBV-infected cells were small to medium size and showed mild to equivocal atypia (Fig. 3A, B). After three months, the lymph nodes of patient 1 were effaced and infiltrated by medium-sized atypical EBV-positive lymphocytes (Fig. 3C-F). On initial examination, the histological findings suggested neoplastic proliferation, but patient 3 displayed polyclonality according to the TCR gene rearrangement (Table 1). The bone marrow of the two patients showed similar findings to the observations in the lymph nodes.

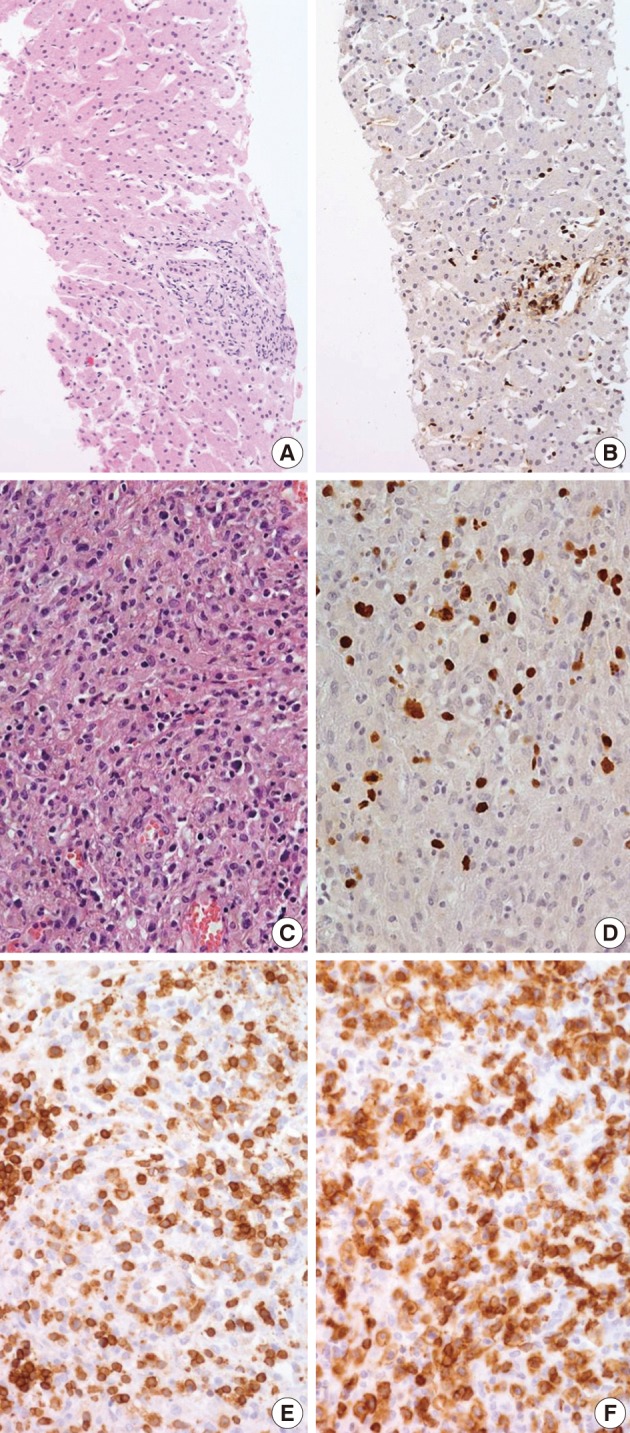
Epstein-Barr virus (EBV)-positive hemophagocytic lymphohistiocytosis (Table 1, patient 3). (A, B) (A) Liver biopsy shows sinusoidal and periportal lymphocytic infiltrates without significant atypia. (B) The majority of lymphocytes are positive for EBV-encoded early RNA (EBER) in situ hybridization. (C, D) After three months from liver biopsy, lymph node biopsy shows infiltration of medium sized atypical lymphocytes with many histiocytes (C). EBER is positive (D). (E, F) Immunohistochemical study reveals that the majority of infiltrated lymphocytes express CD3 (E) and CD8 (F).
The phenotype of the infiltrated cells was CD3-positive, CD20-negative, and CD56-negative in all cases. Systemic T-cell LPD showed mixed CD4- or CD8-positive cells. EBV-positive HLH was CD8-positive T-cell dominant in three patients and CD4-positive T-cell dominant in two patients. In situ hybridization used to detect EBV in biopsy tissues using the EBV-encoded early RNA 1 probe showed the presence of EBV-infected cells in six of seven patients (Fig. 2). In one patient, in situ hybridization failed to detect EBV because the tissue was improperly fixed. The numbers of EBV DNA copies in the peripheral blood varied, ranging up to 5,810 copies/µL.
CAEBV infection
The CAEBV group consisted of 12 patients (eight males and four females), with ages ranging from 10 to 59 years (median, 21 years) (Table 2). Nine patients were children or young adults, one patient was elderly, and their clinical manifestations varied. Eight patients presented with hepatosplenomegaly and lymphadenopathy, and six patients complained of fever. Mosquito-bite hypersensitivity was observed in four children, and three patients had NK lymphocytosis. One child experienced hydroa vacciniforme-like eruption. Some patients manifested unusual clinical findings, such as bowel perforation, immunoglobulin A nephropathy, choreic movement, and brain infarction. Biopsies of the bone marrow, skin, gastrointestinal tract, or lung were performed. The histology varied according to the site of biopsy. EBV-positive lymphocytes were scattered, and infiltrated lymphocytes were predominantly small and devoid of atypia (Fig. 4). Hemophagocytic histiocytosis was found in seven of 11 bone marrow tissues. On immunohistochemistry, CD4-positive T-cells were dominant in three patients and CD8-positive cells were dominant in three patients.
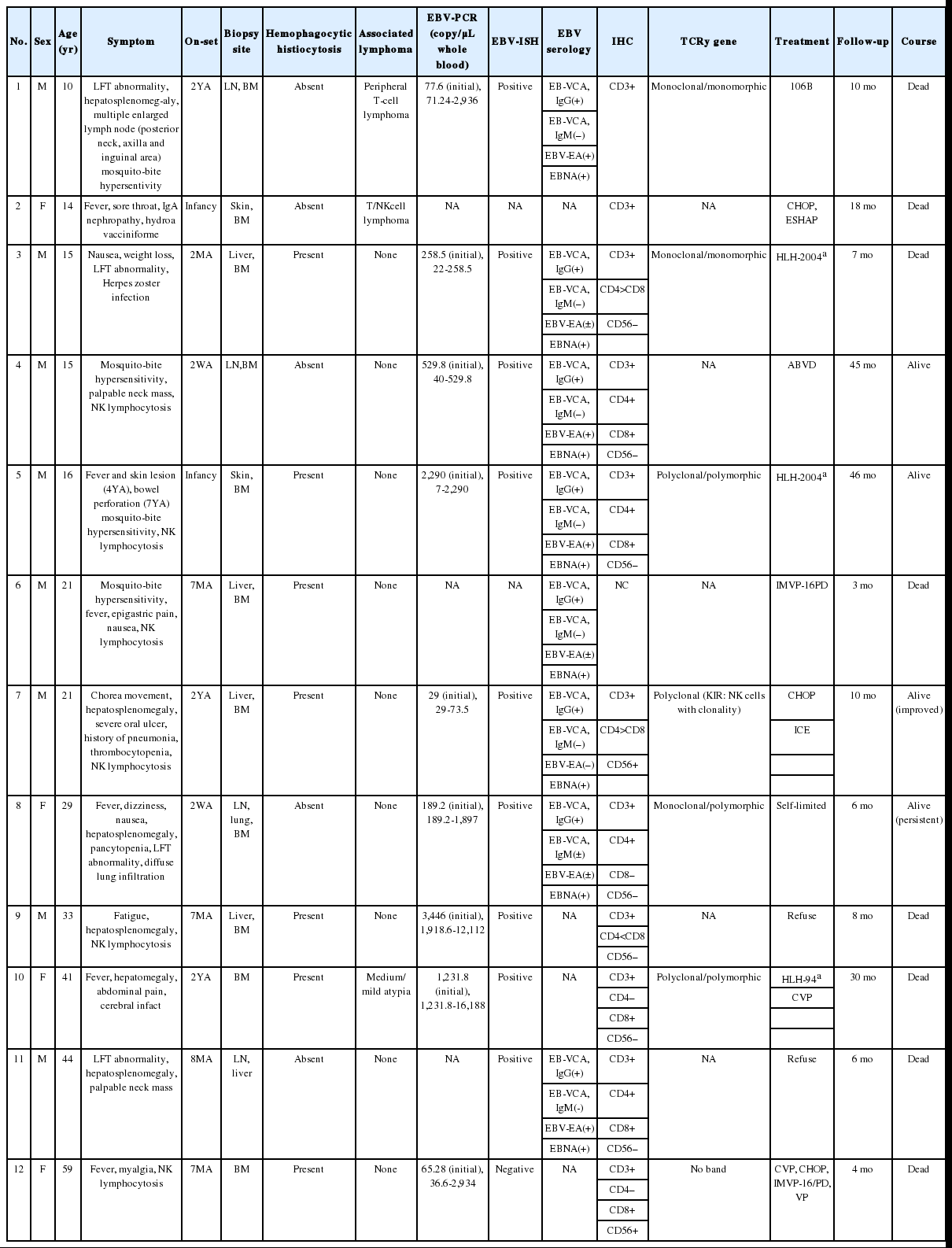
Clinicopathological findings of CAEBV infection
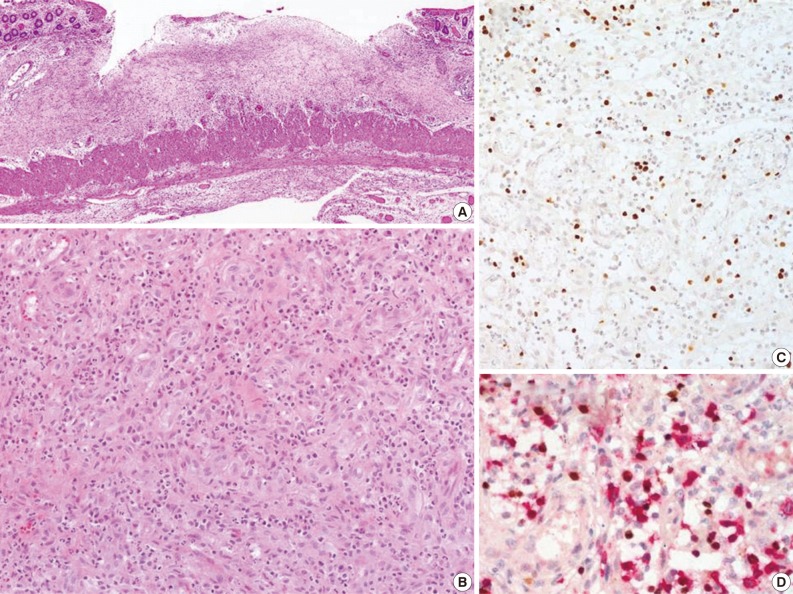
Chronic active Epstein-Barr virus (EBV) infection (Table 2, patient 5). (A) Perforated small intestine shows ulceration with granulation tissue. (B) Infiltrated cells are acute inflammatory cells and small lymphocytes without atypia. (C) EBV-encoded early RNA (EBER) is positive in small lymphocytes. (D) Double procedure for EBER using in situ hybridization (nuclear staining, brown) and immunostaining for CD8 (cytoplasmic staining, red) show EBER-positive cytotoxic T lymphocytes.
Monoclonality was detected in three of the six patients in whom PCR analysis of the TCR gene was successful. Among the three polyclonal patients, one was CD56-positive, with a skewed killer cell immunoglobulin-like receptor (KIR) phenotype, suggesting monoclonal NK-cell proliferation. Among the three patients with polyclonal T-cell proliferation, two were alive and one was alive 30 months after diagnosis. Of the three patients with monoclonal T-cell proliferation, two patients with monomorphic histology died of their disease after ten months and seven months, respectively, and one patient with polymorphic histology had persistent disease six months after diagnosis. The median survival was 18 months. Comparison of patient survival according to clonality was not statistically meaningful because of the limited number of patients included in the study, but monoclonal patients tended to have poorer prognosis.
DISCUSSION
EBV-positive HLH is a clinical entity described in the literature that includes a wide spectrum of illnesses, ranging from EBV-associated reactive polyclonal lymphoproliferation to monoclonal disease.10-12 EBV-associated HLH can be divided into three evolutional stages. The innate immune response phase in the first week of EBV infection is followed by the T-cell activation stage in the second week, which is followed later by the macrophage activation stage. During the last evolutionary phase, a substantial percentage of patients may progress to monoclonal T-cell LPD with or without an abnormal karyotype, which is equivalent to EBV-positive systemic T-cell LPD of childhood.13,14 In vitro studies have suggested that the tumor necrosis factor (TNF) receptor secreted by EBV-infected T-cells and the TNF-receptor-associated factors/nuclear factor-κB/extracellular signal-related kinase pathway activated by the EB viral protein, LPM-1, play a role in the progression to T-cell lymphoma.15
The "EBV-positive systemic T-cell LPD of childhood" in the 2008 WHO classification corresponds to the neoplastic stage of EBV-positive HLH. However, the separation of systemic T-cell LPD from EBV-positive HLH based on clonality raises two issues: difficult diagnosis due to the technical limitations of clonality assessment, and the clinical impact of clonality on EBV-positive NK- or T-cell LPD.
The clonality of EBV-infected LPD can be determined by various methods, including TCR gene rearrangement, cytogenetic studies, and investigation of the terminal repeats of the EBV genome. TCR gene rearrangement, detected by conventional PCR analysis or Southern blot hybridization, is inadequate to examine the clonality of T-cell EBV-infected LPD because of its low sensitivity. EBV-infected cells defined as polyclonal by TCR analysis can be monoclonal based on EBV terminal repeat analysis or cytogenetic analysis. The sensitivity of the recently developed BIOMED-2 assay is superior to the conventional PCR assay.16 In addition to low sensitivity, a TCR gene rearrangement study is disadvantageous because it cannot be used for EBV-positive HLH of NK-cell types. EBV usually infects B-cells and uncommonly T-cells, but NK-cells can also be infected. A recent study by Kimura et al.17 demonstrated that a minor proportion of EBV-positive HLH is actually an NK-cell disorder. Human NK-cells use a sophisticated system of inhibitory and stimulatory receptors of the KIR gene family, which are expressed in a clonally distributed manner. NK-cell KIR gene phenotyping can help determine the clonality of NK-cells,18 but fresh samples are required. Thus, this technique has limited utility in the analysis of paraffin-embedded tissues. Analysis of the EBV terminal repeat may be the most sensitive method because it can be applied in T-, B- and NK-cell disorders. However, EBV clonality is difficult to analyze with routine diagnostic biopsies because large amounts of freshly sampled DNA for Southern blot hybridization are required; chromosomal studies share the same issue.
The clonality of EBV-positive HLH has been reported in a few studies from Asia.12,19,20 A study using conventional PCR reported monoclonal TCR gene rearrangements in ten of 30 adult patients and immunoglobulin gene rearrangements in eight of the same 30 patients.19 The study by Imashuku et al.,12 who analyzed the EBV terminal repeat with Southern blotting, revealed a monoclonal pattern in 20 of 24 patients, a biclonal pattern in two patients, and a polyclonal pattern in two patients. In their study, the TCR gene rearrangement was monoclonal in 15 of 25 patients and polyclonal in ten of the same 25 patients. Clonal cytogenetic abnormalities were observed in seven of 23 patients.12 Recently, Matsuda et al.20 analyzed the TCR gene with the BIOMED-2 protocol, the most recent and advanced technique for gene rearrangement studies and found at all six children with HLH showed a clonal band. According to the above-mentioned studies, a significant proportion of EBV-positive HLH is a clonal disease that would be classified as STLPD of childhood by the current WHO classification.
In previous studies that analyzed the clonality of EBV and proliferating cells in EBV-positive HLH, clonality itself apparently had no clinical impact on patient outcome.19 Imashuku et al.12 reported that the clonality of T- and B-cells has no prognostic significance, whereas all patients with cytogenetic abnormalities experience an aggressive clinical course. The clinical significance of a clonal karyotypic abnormality was also reported in other studies.21 During the transformation from benign reactive lymphoid proliferation to overt neoplastic disease, a small clone of EBV-infected T- or NK-cells may emerge11 and transform into more malignant cells as genetic changes accumulate. Early clonal LPD in this neoplastic transformation may show a similar treatment response to polyclonal EBV-positive HLH, but overt malignant lymphoma with chromosomal abnormality will behave differently.
In terms of patient management, the clinician may use chemotherapy as the initial treatment when the patient is diagnosed with systemic T-cell LPD; however, in patients with EBV-positive HLH, the clinical selection of a therapeutic modality depends on the risk factors for EBV-positive HLH: persistently high EBV genome copy number in the serum, acute fulminant or CAEBV infection, abnormal karyotype, and an association with hereditary disease.22,23 Currently, successful therapies such as conservative/mild treatments without etoposide, HLH-94/2004-type treatment with etoposide, and very aggressive lifesaving measures, including hematopoietic stem cell transplantation, are administered to HLH patients,2,3 illustrating a survival rate greater than 75%.24 In our series, one child treated with the HLH-2004 regimen recovered completely and is alive, whereas other patients who received chemotherapy died. Similarly, two patients with STLPD died despite chemotherapy. Because STLPD does not usually respond to chemotherapy, research for new treatments is needed.
CAEBV infection is synonymous with chronic symptomatic EBV infection. After a primary EBV infection, the majority of patients recover completely, but a substantial proportion of patients develop CAEBV infection, which is currently incurable. The clinical activity of the disease may depend on the balance between the host immune function and the virus. The usual symptoms are fever, hepatic dysfunction, lymphadenopathy, or splenomegaly; however, as shown in the present study, patients often present with unusual clinical manifestations, such as chorea, cerebral hemorrhage, generalized myositis, glomerulonephritis, and pulmonary hypertension.25,26 The disease develops in children and adolescents but can also develop in young adults and elderly patients, although with a lower frequency.17,27
CAEBV infection is almost always accompanied by varying degrees of lymphoproliferation. In the Asian population, CAEBV infection mainly involves T- or NK-cells. The clonality of EBV and EBV-infected T- or NK-cells varies and may be polyclonal, oligoclonal, or monoclonal. As the disease progresses from polyclonal lymphoproliferation to monoclonal disease, histological atypia increases.28 Ohshima et al.28 proposed the categorization of CAEBV infection into three groups, polymorphous and polyclonal, polymorphous and monoclonal, or monomorphic and monoclonal, based on the clonality and histological changes. In the series reported by Ohshima et al.,28 eight of 48 patients with CAEBV infection were polyclonal for TCR gene rearrangement, and the infiltrated cells displayed polymorphic histomorphology; 15 patients showed polymorphic morphology and biclonal or monoclonal TCR gene rearrangement; and 25 patients showed monomorphic histomorphology and monoclonal TCR gene rearrangement. Patients with the monomorphic and monoclonal type of CAEBV infection had poorer prognosis than patients with polymorphic polyclonal or polymorphic monoclonal disease. The survival of the polymorphic polyclonal and polymorphic monoclonal groups did not differ significantly.28 According to the 2008 WHO classification, systemic T-cell LPD corresponds to the polymorphic monoclonal and monomorphic monoclonal groups of CAEBV infection. Despite the limitations of a small sample size in our study, the patients with monoclonal CAEBV infection tended to have a poorer prognosis.
In conclusion, EBV-positive HLH and systemic T-cell LPD share similar clinicopathological findings and may constitute a continuous spectrum of acute EBV-associated T- or NK-cell proliferative disorders. The distinction of EBV-positive T-cell LPD from EBV-positive HLH may be difficult during routine diagnoses due to technical limitations of clonality assessment. The clinical impact of clonality in acute EBV-associated T- or NK-cell proliferative disorders is unclear, whereas monoclonal CAEBV infection disease tends to have a worse prognosis.
Notes
No potential conflict of interest relevant to this article was reported.