 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 47(2); 2013 > Article
-
Original Article
EBV-Positive T/NK-Cell Lymphoproliferative Disease of Childhood - Mineui Hong, Young Hyeh Ko, Keon Hee Yoo1, Hong Hoe Koo1, Seok Jin Kim2, Won Seog Kim2, Heejung Park3
-
Korean Journal of Pathology 2013;47(2):137-147.
DOI: https://doi.org/10.4132/KoreanJPathol.2013.47.2.137
Published online: April 24, 2013
Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
1Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
2Department of Hematology-Oncology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
3Department of Pathology, Ewha Womans University Mokdong Hospital, Ewha Womans University School of Medicine, Seoul, Korea.
-
Corresponding Author: Young Hyeh Ko, M.D. Department of Pathology, Samsung Medical Center, Sungkyunkwan University School of Medicine, 81 Irwon-ro, Gangnam-gu, Seoul 135-710, Korea. Tel: +82-2-3410-2762, Fax: +82-2-3410-6398, yhko310@skku.edu
Corresponding Author: Heejung Park, M.D. Department of Pathology, Ewha Womans University Mokdong Hospital, Ewha Womans University School of Medicine, 1071 Anyangcheon-ro, Yangcheon-gu, Seoul 158-710, Korea. Tel: +82-2-2650-5193, Fax: +82-2-2650-2016, zealot096@gmail.com
© 2013 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
-
Background
- Epstein-Barr virus (EBV)-associated hemophagocytic lymphohistiocytosis (HLH), EBV-positive systemic T-cell lymphoproliferative disease (STLPD) of childhood, and chronic active EBV (CAEBV) infection may develop after primary EBV infection. This study reviewed the clinicopathological spectrum of EBV-associated T- and natural killer (NK)-cell LPD, including STLPD and CAEBV infection, with an analysis of T-cell clonality.
-
Methods
- Clinicopathological features of seven patients with EBV-associated HLH or STLPD and 12 patients with CAEBV infection were reviewed. Immunohistochemical staining and a T-cell receptor (TCR) gene rearrangement study were performed.
-
Results
- STLPD and EBV-positive HLH showed significantly overlapping clinicopathological findings. One patient with STLPD and one patient with EBV-positive HLH demonstrated moderate to severe atypia of the infiltrating lymphocytes, whereas the remaining patients lacked significant atypia. Twelve patients had CAEBV infection, four of whom suffered mosquito-bite hypersensitivity, five showed NK lymphocytosis, and one suffered hydroa vacciniforme. Infiltrating lymphocytes were predominantly small and devoid of atypia. Hemophagocytic histiocytosis was found in seven of 11 patients. Monoclonality was detected in three (50%) of the six patients with successful TCR gene analysis.
-
Conclusions
- EBV-positive HLH and STLPD share similar clinicopathological findings and may constitute a continuous spectrum of acute EBV-associated T- or NK-cell proliferative disorders. The distinction of EBV-positive T-cell LPD from EBV-positive HLH may be difficult during routine diagnoses because of the technical limitations of clonality assessment.
- Patients
- CAEBV infection was defined as 1) persistent or recurrent symptoms related to EBV infection for more than three months; 2) high EBV genome levels in affected tissues or peripheral blood; 3) chronic illness that could not be explained by other known disease processes at diagnosis, and 4) no specific underlying immunological abnormality.7
- To identify EBV-positive systemic T-cell LPD of childhood based on the 2008 WHO classification, all patients with EBV-positive hemophagocytic syndrome presenting at our institution were enrolled in the present study. Patient inclusion was based on the fulfillment of the diagnostic criteria for HLH-2004 in addition to the presence of a high viral load in the tissues or peripheral blood.3
- The surgical pathology database of Samsung Medical Center from 1994 to 2012 was screened with keywords including "CAEBV," "EBV," and "hemophagocytosis." Of the 96 cases retrieved using "EBV" and "hemophagocytosis" as keywords, 84 patients were excluded because LPD had arisen in immunocompromised patients, such as those receiving organ transplantation or patients with congenital immune deficiency syndrome. Fifteen patients were diagnosed with CAEBV infection, and 12 patients had paraffin blocks available for examination. Among the remaining 12 patients, seven had paraffin blocks available for examination and were enrolled in the present study. Formalin-fixed paraffin-embedded blocks of liver, bone marrow, and lymph node tissues were used for further investigation.
- Immunohistochemistry
- Immunohistochemical analysis was performed on the paraffin sections using monoclonal and polyclonal antibodies to detect lineage-specific or lineage-characteristic antigens, including CD3 (Dakopatts, Copenhagen, Denmark), CD20 (Novocastra, Newcastle upon Tyne, UK), CD56 (Novocastra), CD4 (Novocastra), and CD8 (Novocastra).
- T-cell receptor (TCR) gene rearrangement
- For the polymerase chain reaction (PCR) amplification of the TCR γ locus, DNA was extracted from the paraffin-embedded formalin-fixed tissues with the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). Conventional PCR followed by single-stranded conformational polymorphism analysis was performed, as previously described.8 When conventional PCR for TCR γ genes produced negative results, further analysis was performed using the TCR β and γ primers, according to the recommendations of the BIOMED-2 protocols (Invivoscribe, San Diego, CA, USA).9
MATERIALS AND METHODS
- All patients (12 males and eight females) were Korean, ranging from 4 to 61 years of age.
- EBV-positive hemophagocytic syndrome
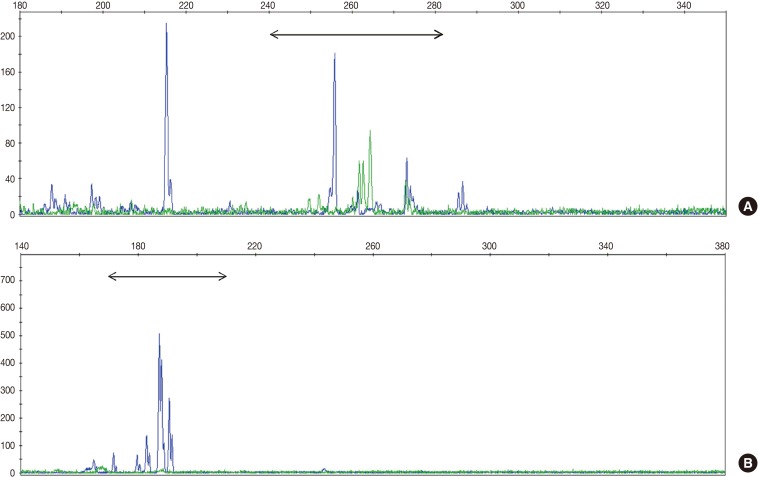
- TCR gene rearrangement analysis revealed monoclonality in two patients (28.6%) and polyclonality in four patients (57.1%) (Fig. 1). The analysis of one patient failed because the DNA quality was poor. Two patients with T-cell monoclonality were classified as suffering systemic T-cell LPD and the remaining patients as suffering EBV-positive HLH.
- Of the two patients with systemic T-cell LPD, one was a young female and the other an adult male. The EBV-positive HLH patients included two children, two young adults, and one elderly adult, with ages ranging from 4 to 61 years (median, 20 years). The clinicopathological findings for the two conditions were similar, and almost all patients had acute-onset fever. Their symptoms included lymph node enlargement and hepatosplenomegaly. The duration of the symptoms was two months in patients with systemic T-cell LPD and ranged from three days to one month (median, 1 month) in EBV-positive HLH patients. Two patients with systemic T-cell LPD received chemotherapy but died after two and three months, respectively. Among the five patients with EBV-positive HLH, four received chemotherapy or steroid and died after a median period of one month, whereas the one patient treated with the HLH-2004 regimen recovered completely and was alive at the final follow-up. The EBV load in the peripheral blood was analyzed using whole blood, and the loads during the clinical course are shown in Table 1. The levels were variable, and two patients with EBV-positive HLH showed high viral loads in their peripheral blood.
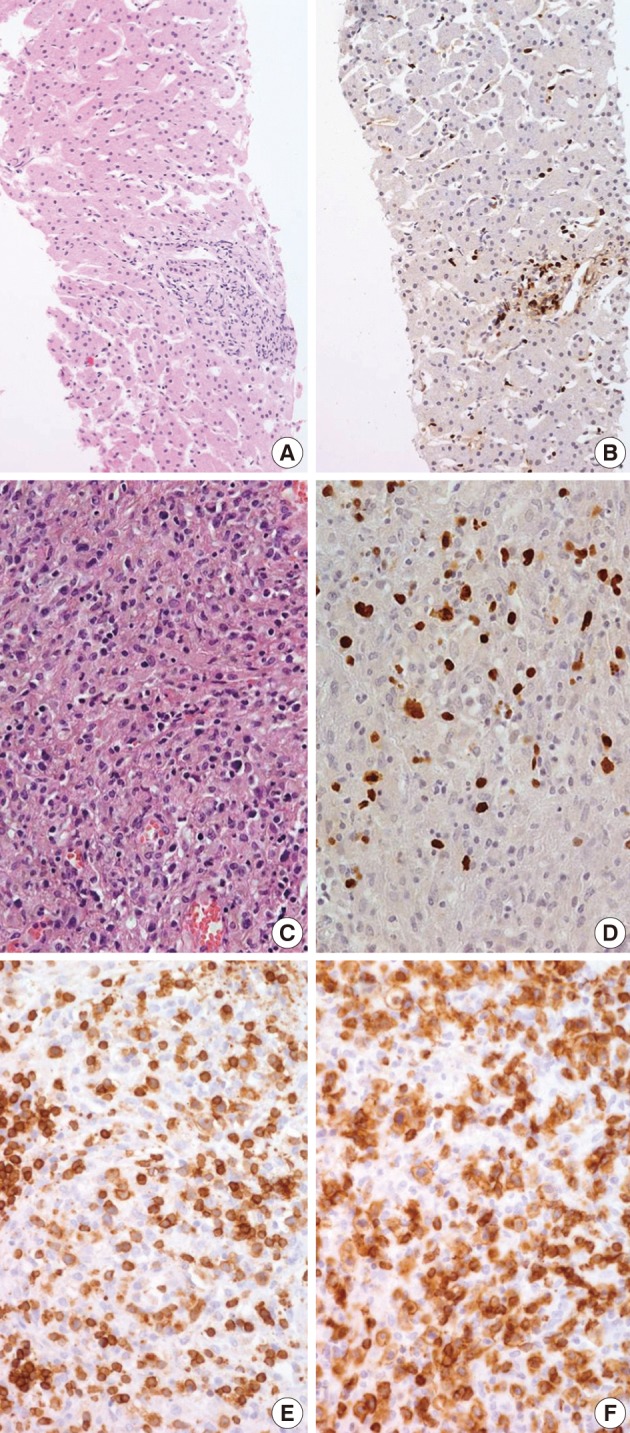
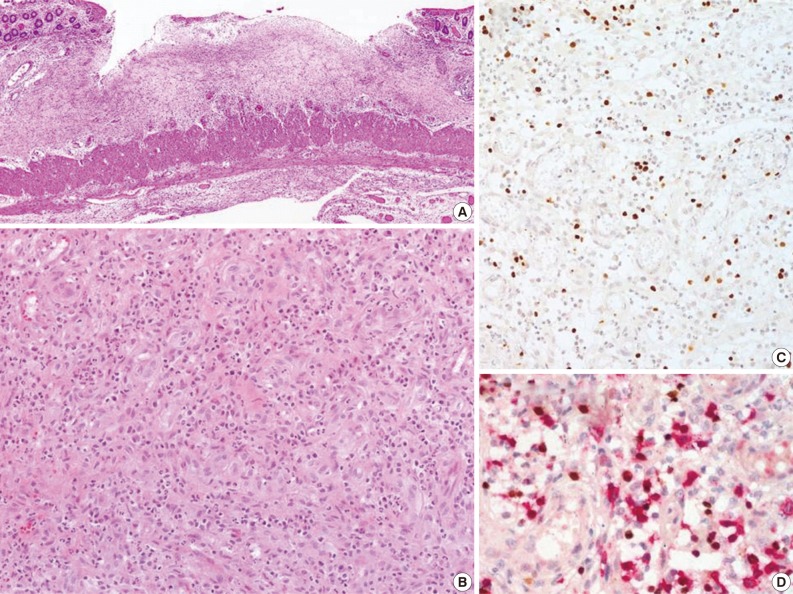
- Biopsies were obtained from the bone marrow of six patients, lymph nodes of two patients, liver of one patient, and spleen of one patient. In the bone marrow, liver, and spleen samples, the number of lymphocytes was slightly increased and showed mild to equivocal atypia. Hemophagocytic histiocytosis was observed in all patients. The lymph node biopsies from patient 1 (systemic T-cell LPD) and patient 3 (EBV-positive HLH) demonstrated diffuse effacement of the nodal architecture by the infiltration of monotonous medium-sized atypical lymphocytes (Fig. 2). The lymphocytes had hyperchromatic nuclei and irregular nuclear contours. At the first liver biopsy of patient 3, EBV-infected cells were small to medium size and showed mild to equivocal atypia (Fig. 3A, B). After three months, the lymph nodes of patient 1 were effaced and infiltrated by medium-sized atypical EBV-positive lymphocytes (Fig. 3C-F). On initial examination, the histological findings suggested neoplastic proliferation, but patient 3 displayed polyclonality according to the TCR gene rearrangement (Table 1). The bone marrow of the two patients showed similar findings to the observations in the lymph nodes.
- The phenotype of the infiltrated cells was CD3-positive, CD20-negative, and CD56-negative in all cases. Systemic T-cell LPD showed mixed CD4- or CD8-positive cells. EBV-positive HLH was CD8-positive T-cell dominant in three patients and CD4-positive T-cell dominant in two patients. In situ hybridization used to detect EBV in biopsy tissues using the EBV-encoded early RNA 1 probe showed the presence of EBV-infected cells in six of seven patients (Fig. 2). In one patient, in situ hybridization failed to detect EBV because the tissue was improperly fixed. The numbers of EBV DNA copies in the peripheral blood varied, ranging up to 5,810 copies/µL.
- CAEBV infection
- The CAEBV group consisted of 12 patients (eight males and four females), with ages ranging from 10 to 59 years (median, 21 years) (Table 2). Nine patients were children or young adults, one patient was elderly, and their clinical manifestations varied. Eight patients presented with hepatosplenomegaly and lymphadenopathy, and six patients complained of fever. Mosquito-bite hypersensitivity was observed in four children, and three patients had NK lymphocytosis. One child experienced hydroa vacciniforme-like eruption. Some patients manifested unusual clinical findings, such as bowel perforation, immunoglobulin A nephropathy, choreic movement, and brain infarction. Biopsies of the bone marrow, skin, gastrointestinal tract, or lung were performed. The histology varied according to the site of biopsy. EBV-positive lymphocytes were scattered, and infiltrated lymphocytes were predominantly small and devoid of atypia (Fig. 4). Hemophagocytic histiocytosis was found in seven of 11 bone marrow tissues. On immunohistochemistry, CD4-positive T-cells were dominant in three patients and CD8-positive cells were dominant in three patients.
- Monoclonality was detected in three of the six patients in whom PCR analysis of the TCR gene was successful. Among the three polyclonal patients, one was CD56-positive, with a skewed killer cell immunoglobulin-like receptor (KIR) phenotype, suggesting monoclonal NK-cell proliferation. Among the three patients with polyclonal T-cell proliferation, two were alive and one was alive 30 months after diagnosis. Of the three patients with monoclonal T-cell proliferation, two patients with monomorphic histology died of their disease after ten months and seven months, respectively, and one patient with polymorphic histology had persistent disease six months after diagnosis. The median survival was 18 months. Comparison of patient survival according to clonality was not statistically meaningful because of the limited number of patients included in the study, but monoclonal patients tended to have poorer prognosis.
RESULTS
TCR gene rearrangement
Clinicopathological findings of systemic T-cell LPD and EBV-positive HLH
- EBV-positive HLH is a clinical entity described in the literature that includes a wide spectrum of illnesses, ranging from EBV-associated reactive polyclonal lymphoproliferation to monoclonal disease.10-12 EBV-associated HLH can be divided into three evolutional stages. The innate immune response phase in the first week of EBV infection is followed by the T-cell activation stage in the second week, which is followed later by the macrophage activation stage. During the last evolutionary phase, a substantial percentage of patients may progress to monoclonal T-cell LPD with or without an abnormal karyotype, which is equivalent to EBV-positive systemic T-cell LPD of childhood.13,14 In vitro studies have suggested that the tumor necrosis factor (TNF) receptor secreted by EBV-infected T-cells and the TNF-receptor-associated factors/nuclear factor-κB/extracellular signal-related kinase pathway activated by the EB viral protein, LPM-1, play a role in the progression to T-cell lymphoma.15
- The "EBV-positive systemic T-cell LPD of childhood" in the 2008 WHO classification corresponds to the neoplastic stage of EBV-positive HLH. However, the separation of systemic T-cell LPD from EBV-positive HLH based on clonality raises two issues: difficult diagnosis due to the technical limitations of clonality assessment, and the clinical impact of clonality on EBV-positive NK- or T-cell LPD.
- The clonality of EBV-infected LPD can be determined by various methods, including TCR gene rearrangement, cytogenetic studies, and investigation of the terminal repeats of the EBV genome. TCR gene rearrangement, detected by conventional PCR analysis or Southern blot hybridization, is inadequate to examine the clonality of T-cell EBV-infected LPD because of its low sensitivity. EBV-infected cells defined as polyclonal by TCR analysis can be monoclonal based on EBV terminal repeat analysis or cytogenetic analysis. The sensitivity of the recently developed BIOMED-2 assay is superior to the conventional PCR assay.16 In addition to low sensitivity, a TCR gene rearrangement study is disadvantageous because it cannot be used for EBV-positive HLH of NK-cell types. EBV usually infects B-cells and uncommonly T-cells, but NK-cells can also be infected. A recent study by Kimura et al.17 demonstrated that a minor proportion of EBV-positive HLH is actually an NK-cell disorder. Human NK-cells use a sophisticated system of inhibitory and stimulatory receptors of the KIR gene family, which are expressed in a clonally distributed manner. NK-cell KIR gene phenotyping can help determine the clonality of NK-cells,18 but fresh samples are required. Thus, this technique has limited utility in the analysis of paraffin-embedded tissues. Analysis of the EBV terminal repeat may be the most sensitive method because it can be applied in T-, B- and NK-cell disorders. However, EBV clonality is difficult to analyze with routine diagnostic biopsies because large amounts of freshly sampled DNA for Southern blot hybridization are required; chromosomal studies share the same issue.
- The clonality of EBV-positive HLH has been reported in a few studies from Asia.12,19,20 A study using conventional PCR reported monoclonal TCR gene rearrangements in ten of 30 adult patients and immunoglobulin gene rearrangements in eight of the same 30 patients.19 The study by Imashuku et al.,12 who analyzed the EBV terminal repeat with Southern blotting, revealed a monoclonal pattern in 20 of 24 patients, a biclonal pattern in two patients, and a polyclonal pattern in two patients. In their study, the TCR gene rearrangement was monoclonal in 15 of 25 patients and polyclonal in ten of the same 25 patients. Clonal cytogenetic abnormalities were observed in seven of 23 patients.12 Recently, Matsuda et al.20 analyzed the TCR gene with the BIOMED-2 protocol, the most recent and advanced technique for gene rearrangement studies and found at all six children with HLH showed a clonal band. According to the above-mentioned studies, a significant proportion of EBV-positive HLH is a clonal disease that would be classified as STLPD of childhood by the current WHO classification.
- In previous studies that analyzed the clonality of EBV and proliferating cells in EBV-positive HLH, clonality itself apparently had no clinical impact on patient outcome.19 Imashuku et al.12 reported that the clonality of T- and B-cells has no prognostic significance, whereas all patients with cytogenetic abnormalities experience an aggressive clinical course. The clinical significance of a clonal karyotypic abnormality was also reported in other studies.21 During the transformation from benign reactive lymphoid proliferation to overt neoplastic disease, a small clone of EBV-infected T- or NK-cells may emerge11 and transform into more malignant cells as genetic changes accumulate. Early clonal LPD in this neoplastic transformation may show a similar treatment response to polyclonal EBV-positive HLH, but overt malignant lymphoma with chromosomal abnormality will behave differently.
- In terms of patient management, the clinician may use chemotherapy as the initial treatment when the patient is diagnosed with systemic T-cell LPD; however, in patients with EBV-positive HLH, the clinical selection of a therapeutic modality depends on the risk factors for EBV-positive HLH: persistently high EBV genome copy number in the serum, acute fulminant or CAEBV infection, abnormal karyotype, and an association with hereditary disease.22,23 Currently, successful therapies such as conservative/mild treatments without etoposide, HLH-94/2004-type treatment with etoposide, and very aggressive lifesaving measures, including hematopoietic stem cell transplantation, are administered to HLH patients,2,3 illustrating a survival rate greater than 75%.24 In our series, one child treated with the HLH-2004 regimen recovered completely and is alive, whereas other patients who received chemotherapy died. Similarly, two patients with STLPD died despite chemotherapy. Because STLPD does not usually respond to chemotherapy, research for new treatments is needed.
- CAEBV infection is synonymous with chronic symptomatic EBV infection. After a primary EBV infection, the majority of patients recover completely, but a substantial proportion of patients develop CAEBV infection, which is currently incurable. The clinical activity of the disease may depend on the balance between the host immune function and the virus. The usual symptoms are fever, hepatic dysfunction, lymphadenopathy, or splenomegaly; however, as shown in the present study, patients often present with unusual clinical manifestations, such as chorea, cerebral hemorrhage, generalized myositis, glomerulonephritis, and pulmonary hypertension.25,26 The disease develops in children and adolescents but can also develop in young adults and elderly patients, although with a lower frequency.17,27
- CAEBV infection is almost always accompanied by varying degrees of lymphoproliferation. In the Asian population, CAEBV infection mainly involves T- or NK-cells. The clonality of EBV and EBV-infected T- or NK-cells varies and may be polyclonal, oligoclonal, or monoclonal. As the disease progresses from polyclonal lymphoproliferation to monoclonal disease, histological atypia increases.28 Ohshima et al.28 proposed the categorization of CAEBV infection into three groups, polymorphous and polyclonal, polymorphous and monoclonal, or monomorphic and monoclonal, based on the clonality and histological changes. In the series reported by Ohshima et al.,28 eight of 48 patients with CAEBV infection were polyclonal for TCR gene rearrangement, and the infiltrated cells displayed polymorphic histomorphology; 15 patients showed polymorphic morphology and biclonal or monoclonal TCR gene rearrangement; and 25 patients showed monomorphic histomorphology and monoclonal TCR gene rearrangement. Patients with the monomorphic and monoclonal type of CAEBV infection had poorer prognosis than patients with polymorphic polyclonal or polymorphic monoclonal disease. The survival of the polymorphic polyclonal and polymorphic monoclonal groups did not differ significantly.28 According to the 2008 WHO classification, systemic T-cell LPD corresponds to the polymorphic monoclonal and monomorphic monoclonal groups of CAEBV infection. Despite the limitations of a small sample size in our study, the patients with monoclonal CAEBV infection tended to have a poorer prognosis.
- In conclusion, EBV-positive HLH and systemic T-cell LPD share similar clinicopathological findings and may constitute a continuous spectrum of acute EBV-associated T- or NK-cell proliferative disorders. The distinction of EBV-positive T-cell LPD from EBV-positive HLH may be difficult during routine diagnoses due to technical limitations of clonality assessment. The clinical impact of clonality in acute EBV-associated T- or NK-cell proliferative disorders is unclear, whereas monoclonal CAEBV infection disease tends to have a worse prognosis.
DISCUSSION
- 1. Rickinson AB. Chronic, symptomatic Epstein-Barr virus infections. Immunol Today 1986; 7: 13-14. ArticlePubMed
- 2. Henter JI, Aricò M, Egeler RM, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol 1997; 28: 342-347. ArticlePubMed
- 3. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48: 124-131. ArticlePubMed
- 4. Quintanilla-Martinez L, Kimura H, Jaffe ES. In: Swerdlow SH, Campo E, Harris NL, eds. EBV-positive T-cell lymphoproliferative disorders of childhood. WHO classification of tumours of haematopoietic and lymphoid tissues. 2008; 4th ed. Lyon: IARC Press, 278-280.
- 5. Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood 2000; 96: 443-451. ArticlePubMedPDF
- 6. Kimura H, Morishima T, Kanegane H, et al. Prognostic factors for chronic active Epstein-Barr virus infection. J Infect Dis 2003; 187: 527-533. ArticlePubMed
- 7. Okano M, Kawa K, Kimura H, et al. Proposed guidelines for diagnosing chronic active Epstein-Barr virus infection. Am J Hematol 2005; 80: 64-69. ArticlePubMed
- 8. Signoretti S, Murphy M, Cangi MG, Puddu P, Kadin ME, Loda M. Detection of clonal T-cell receptor gamma gene rearrangements in paraffin-embedded tissue by polymerase chain reaction and nonradioactive single-strand conformational polymorphism analysis. Am J Pathol 1999; 154: 67-75. PubMedPMC
- 9. van Dongen JJ, Langerak AW, Brüggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003; 17: 2257-2317. ArticlePubMedPDF
- 10. Wada T, Kurokawa T, Toma T, et al. Immunophenotypic analysis of Epstein-Barr virus (EBV)-infected CD8(+) T cells in a patient with EBV-associated hemophagocytic lymphohistiocytosis. Eur J Haematol 2007; 79: 72-75. ArticlePubMed
- 11. Lin MT, Chang HM, Huang CJ, et al. Massive expansion of EBV+ monoclonal T cells with CD5 down regulation in EBV-associated haemophagocytic lymphohistiocytosis. J Clin Pathol 2007; 60: 101-103. ArticlePubMedPMC
- 12. Imashuku S, Hibi S, Tabata Y, et al. Outcome of clonal hemophagocytic lymphohistiocytosis: analysis of 32 cases. Leuk Lymphoma 2000; 37: 577-584. ArticlePubMed
- 13. Su IJ, Wang CH, Cheng AL, Chen RL. Hemophagocytic syndrome in Epstein-Barr virus-associated T-lymphoproliferative disorders: disease spectrum, pathogenesis, and management. Leuk Lymphoma 1995; 19: 401-406. ArticlePubMed
- 14. Chen RL, Su IJ, Lin KH, et al. Fulminant childhood hemophagocytic syndrome mimicking histiocytic medullary reticulosis: an atypical form of Epstein-Barr virus infection. Am J Clin Pathol 1991; 96: 171-176. ArticlePubMed
- 15. Chuang HC, Lay JD, Hsieh WC, Su IJ. Pathogenesis and mechanism of disease progression from hemophagocytic lymphohistiocytosis to Epstein-Barr virus-associated T-cell lymphoma: nuclear factor-kappa B pathway as a potential therapeutic target. Cancer Sci 2007; 98: 1281-1287. PubMedPMC
- 16. Liu H, Bench AJ, Bacon CM, et al. A practical strategy for the routine use of BIOMED-2 PCR assays for detection of B- and T-cell clonality in diagnostic haematopathology. Br J Haematol 2007; 138: 31-43. ArticlePubMed
- 17. Kimura H, Ito Y, Kawabe S, et al. EBV-associated T/NK-cell lymphoproliferative diseases in nonimmunocompromised hosts: prospective analysis of 108 cases. Blood 2012; 119: 673-686. ArticlePubMedPDF
- 18. Sawada A, Sato E, Koyama M, et al. NK-cell repertoire is feasible for diagnosing Epstein-Barr virus-infected NK-cell lymphoproliferative disease and evaluating the treatment effect. Am J Hematol 2006; 81: 576-581. ArticlePubMed
- 19. Ahn JS, Rew SY, Shin MG, et al. Clinical significance of clonality and Epstein-Barr virus infection in adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol 2010; 85: 719-722. ArticlePubMed
- 20. Matsuda K, Nakazawa Y, Yanagisawa R, Honda T, Ishii E, Koike K. Detection of T-cell receptor gene rearrangement in children with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis using the BIOMED-2 multiplex polymerase chain reaction combined with GeneScan analysis. Clin Chim Acta 2011; 412: 1554-1558. ArticlePubMed
- 21. Ito E, Kitazawa J, Arai K, et al. Fatal Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis with clonal karyotype abnormality. Int J Hematol 2000; 71: 263-265. PubMed
- 22. Imashuku S. Treatment of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis (EBV-HLH); update 2010. J Pediatr Hematol Oncol 2011; 33: 35-39. ArticlePubMed
- 23. Imashuku S, Kuriyama K, Sakai R, et al. Treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH) in young adults: a report from the HLH study center. Med Pediatr Oncol 2003; 41: 103-109. ArticlePubMed
- 24. Imashuku S, Teramura T, Tauchi H, et al. Longitudinal follow-up of patients with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Haematologica 2004; 89: 183-188. PubMed
- 25. Kano K, Yamada Y, Sato Y, Arisaka O, Ono Y, Ueda Y. Glomerulonephritis in a patient with chronic active Epstein-Barr virus infection. Pediatr Nephrol 2005; 20: 89-92. ArticlePubMedPDF
- 26. Uchiyama T, Arai K, Yamamoto-Tabata T, et al. Generalized myositis mimicking polymyositis associated with chronic active Epstein-Barr virus infection. J Neurol 2005; 252: 519-525. ArticlePubMedPDF
- 27. Isobe Y, Aritaka N, Setoguchi Y, et al. T/NK cell type chronic active Epstein-Barr virus disease in adults: an underlying condition for Epstein-Barr virus-associated T/NK-cell lymphoma. J Clin Pathol 2012; 65: 278-282. ArticlePubMed
- 28. Ohshima K, Kimura H, Yoshino T, et al. Proposed categorization of pathological states of EBV-associated T/natural killer-cell lymphoproliferative disorder (LPD) in children and young adults: overlap with chronic active EBV infection and infantile fulminant EBV T-LPD. Pathol Int 2008; 58: 209-217. ArticlePubMed
REFERENCES

| No. | Sex | Age (yr) | Symptom | On-set | Biopsy site | Hemophagocytic histiocytosis | Cell size/Atypia | EBV-PCR (copy/μL whole blood) | EBV-ISH | EBV serology | IHC | TCRy gene | Treatment | Follow-up | Course |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Systemic T-cell LPD | |||||||||||||||
| 1 | F | 7 | Fever, cervical lymph node enlargement | 2MA | LN,BM | Present | Medium/moderate atypia | 42 (initial), 2.5-42 | Positive | EB-VCA, lgG(+) EB- | CD3+ | Monoclonal | VHR, L-Asp | 3 mo | Dead |
| VCA, IgM(-) | CD4<CD8 | ||||||||||||||
| EBV-EA(+) | CD56- | ||||||||||||||
| EBNA(+) | |||||||||||||||
| 2 | M | 43 | Fever, splenomegaly | 2MA | BM | Present | Small/mild atypia | 142 (initial), 0-142 | Positive | NC | CD3+ | Monoclonal | IMVP-16/PD | 2 mo | Dead |
| CD4+ | |||||||||||||||
| CD8+ | |||||||||||||||
| CD56– | |||||||||||||||
| EBV-positive hemophagocytic lymphohistiocytosis | |||||||||||||||
| 3 | F | 4 | Fever, jaundice, hepatomegaly, leukocytopenia | 2WA | LN, BM, liver | Present | Medium/severe atypia | 506 (initial), 4.77-6,422 | Positive | EB-VCA, igG(+) | CD3+ | Polyclonal | 106B, VHR | 3 mo | Dead |
| EB-VCA, lgM(+) | CD4- | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(-) | CD56– | ||||||||||||||
| 4 | M | 10 | Cervical lymph node enlargement, persistent fever, hepatosplenomegaly, LFT abnormality, pancytopenia, facial petechiae, gingival swelling | 1MA | BM | Present | Medium/moderate atypia | 5,810 (initial), 8.4-5, 810 | Failed | NA | CD3+ | No band | HLH-2004a | 69 mo | Alive |
| CD4- | |||||||||||||||
| CD8+ | |||||||||||||||
| CD56- | |||||||||||||||
| 5 | F | 20 | Headache, fever, night sweat, abdominal pain, hepatosplenomegaly | 3DA | BM | Present | Small/mild atypia | 8.5 | Positive | NA | CD3+ | Polyclonal | CHOP | 1 mo | Dead |
| CD4<CD8 | |||||||||||||||
| CD56– | |||||||||||||||
| 6 | M | 33 | Fever | 1MA | Liver | Present | Small/mild atypia | 105.7 | Positive | NA | CD3+ | Polyclonal | Steroid | 3 days | Dead |
| CD4+CD8+ | |||||||||||||||
| CD56- | |||||||||||||||
| 7 | F | 61 | Fever, chill | 4WA | Spleen, BM | Present | Small/mild atypia | NA | Positive | EB-VCA, igG(+) | CD3+ | Polyclonal | Steroid | 1 mo | Dead |
| EB-VCA, lgM(-) | CD4+ | ||||||||||||||
| EBV-EA(-) | CDS- | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
Treatment regimen; VHR: prednisolone, cyclophosphamide, daunorubicin, vincristine, L-asparaginase, intrathecal methotrexate, L-Asp: L-asparaginase, IMVP-16/PD: ifosfamide, methotrexate, etoposide, prednisolone, 106B: prednisolone, cyclophosphamide, daunorubicin, vincristine, L-asparaginase, CHOP: cyclophosphamide, doxorubicin, vincristine, prednisolone.
LPD, lymphoproliferative disease; EBV, Epstein-Barr virus; PCR, polymerase chain reaction; ISH, in situ hybridization; IHC, immunohistochemistry; TCR, T-cell receptor; F, female; MA, months ago; LN, lymph node; BM, bone marrow; EB-VCA, EB viral capsid antigen; EBV-EA, EBV early antigen complex; EBNA, EBV nuclear antigen; M, male; WA, weeks ago; LFT, liver function test; NA, not available; DA, days ago.
aHLH-94/2004: dexamethasone, cyclosporinA, intravenous Ig.
| No. | Sex | Age (yr) | Symptom | On-set | Biopsy site | Hemophagocytic histiocytosis | Associated lymphoma | EBV-PCR (copy/μL whole blood) | EBV-ISH | EBV serology | IHC | TCRy gene | Treatment | Follow-up | Course |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 10 | LFT abnormality, hepatosplenomeg-aly, multiple enlarged lymph node (posterior neck, axilla and inguinal area) mosquito-bite hypersentivity | 2YA | LN, BM | Absent | Peripheral T-cell lymphoma | 77.6 (initial), 71.24-2,936 | Positive | EB-VCA, IgG(+) | CD3+ | Monoclonal/monomorphic | 106B | 10 mo | Dead |
| EB-VCA, IgM(–) | |||||||||||||||
| EBV-EA(+) | |||||||||||||||
| EBNA(+) | |||||||||||||||
| 2 | F | 14 | Fever, sore throat, IgA nephropathy, hydroa vacciniforme | Infancy | Skin, BM | Absent | T/NKcell lymphoma | NA | NA | NA | CD3+ | NA | CHOP, ESHAP | 18 mo | Dead |
| 3 | M | 15 | Nausea, weight loss, LFT abnormality, Herpes zoster infection | 2MA | Liver, BM | Present | None | 258.5 (initial), 22-258.5 | Positive | EB-VCA, IgG(+) | CD3+ | Monoclonal/monomorphic | HLH-2004a | 7 mo | Dead |
| EB-VCA, IgM(–) | CD4>CD8 | ||||||||||||||
| EBV-EA(±) | CD56– | ||||||||||||||
| EBNA(+) | |||||||||||||||
| 4 | M | 15 | Mosquito-bite hypersensitivity, palpable neck mass, NK lymphocytosis | 2WA | LN,BM | Absent | None | 529.8 (initial), 40-529.8 | Positive | EB-VCA, IgG(+) | CD3+ | NA | ABVD | 45 mo | Alive |
| EB-VCA, IgM(–) | CD4+ | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 5 | M | 16 | Fever and skin lesion (4YA), bowel perforation (7YA) mosquito-bite hypersensitivity, NK lymphocytosis | Infancy | Skin, BM | Present | None | 2,290 (initial), 7-2,290 | Positive | EB-VCA, IgG(+) | CD3+ | Polyclonal/polymorphic | HLH-2004a | 46 mo | Alive |
| EB-VCA, IgM(–) | CD4+ | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 6 | M | 21 | Mosquito-bite hypersensitivity, fever, epigastric pain, nausea, NK lymphocytosis | 7MA | Liver, BM | Present | None | NA | NA | EB-VCA, IgG(+) | NC | NA | IMVP-16PD | 3 mo | Dead |
| EB-VCA, IgM(–) | |||||||||||||||
| EBV-EA(±) | |||||||||||||||
| EBNA(+) | |||||||||||||||
| 7 | M | 21 | Chorea movement, hepatosplenomegaly, severe oral ulcer, history of pneumonia, thrombocytopenia, NK lymphocytosis | 2YA | Liver, BM | Present | None | 29 (initial), 29-73.5 | Positive | EB-VCA, IgG(+) | CD3+ | Polyclonal (KIR: NK cells with clonality) | CHOP | 10 mo | Alive (improved) |
| EB-VCA, IgM(–) | CD4>CD8 | ICE | |||||||||||||
| EBV-EA(–) | CD56+ | ||||||||||||||
| EBNA(+) | |||||||||||||||
| 8 | F | 29 | Fever, dizziness, nausea, hepatosplenomegaly, pancytopenia, LFT abnormality, diffuse lung infiltration | 2WA | LN, lung, BM | Absent | None | 189.2 (initial), 189.2-1,897 | Positive | EB-VCA, IgG(+) | CD3+ | Monoclonal/polymorphic | Self-limited | 6 mo | Alive (persistent) |
| EB-VCA, IgM(±) | CD4+ | ||||||||||||||
| EBV-EA(±) | CD8– | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 9 | M | 33 | Fatigue, hepatosplenomegaly, NK lymphocytosis | 7MA | Liver, BM | Present | None | 3,446 (initial), 1,918.6-12,112 | Positive | NA | CD3+ | NA | Refuse | 8 mo | Dead |
| CD4<CD8 | |||||||||||||||
| CD56– | |||||||||||||||
| 10 | F | 41 | Fever, hepatomegaly, abdominal pain, cerebral infact | 2YA | BM | Present | Medium/ mild atypia | 1,231.8 (initial), 1,231.8-16,188 | Positive | NA | CD3+ | Polyclonal/polymorphic | HLH-94a | 30 mo | Dead |
| CD4– | CVP | ||||||||||||||
| CD8+ | |||||||||||||||
| CD56– | |||||||||||||||
| 11 | M | 44 | LFT abnormality, hepatosplenomegaly, palpable neck mass | 8MA | LN, liver | Absent | None | NA | Positive | EB-VCA, IgG(+) | CD3+ | NA | Refuse | 6 mo | Dead |
| EB-VCA, IgM(-) | CD4+ | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 12 | F | 59 | Fever, myalgia, NK lymphocytosis | 7MA | BM | Present | None | 65.28 (initial), 36.6-2,934 | Negative | NA | CD3+ | No band | CVP, CHOP, IMVP-16/PD, VP | 4 mo | Dead |
| CD4– | |||||||||||||||
| CD8+ | |||||||||||||||
| CD56+ |
Treatment regimen; 106B: prednisolone, cyclophosphamide, daunorubicin, vincristine, L-asparaginase, CHOP: cyclophosphamide, daunorubicin, vincristine, prednisolone, ESHAP: etoposide, methylprednisolone, high-dose cytarabine, cisplatin, ABVD: adriamycin, bleomycin, vinblastine, dacarbazine, IMVP-16/PD: ifosfamide, methotrexate, etoposide, prednisolone, ICE: ifosfamide, carboplatin, etoposide, CVP: cyclophosphamide, vincristine, prednisolone.
CAEBV, chronic active Epstein-Barr virus; EBV, Epstein-Barr virus; PCR, polymerase chain reaction; ISH, in situ hybridization; IHC, immunohistochemistry; TCR, T-cell receptor; M, male; LFT, liver function test; YA, years ago; LN, lymph node; BM, bone marrow; EB-VCA, EB viral capsid antigen; EBV-EA, EBV early antigen complex; EBNA, EBV nuclear antigen; F, female; NK, natural killer; NA, not available; MA, months ago; WA, weeks ago; KIR, killer cell immunoglobulin-like receptor.
aHLH-94/2004: dexamethasone, cyclosporinA, intravenous Ig.
Figure & Data
References
Citations
- Chronic active Epstein-Barr virus disease: molecular pathogenesis, evolving concepts, and emerging therapies
Hiroshi Kimura, Jeffrey I. Cohen
Blood.2026; 147(14): 1562. CrossRef - Histopathological characteristics of Epstein-Barr virus (EBV)–associated encephalitis and colitis in chronic active EBV infection
Betty A Kasimo, James J Yahaya, Sun Och Yoon, Se Hoon Kim, Minsun Jung
Journal of Pathology and Translational Medicine.2025; 59(3): 188. CrossRef - Die fünfte Auflage der WHO‐Klassifikation – Was ist neu für kutane Lymphome?
Susanne Melchers, Jana D. Albrecht, Werner Kempf, Jan P. Nicolay
JDDG: Journal der Deutschen Dermatologischen Gesellschaft.2024; 22(9): 1254. CrossRef - Fifth Edition of the World Health Organization Classification of Tumors of the Hematopoietic and Lymphoid Tissues: Mature T-Cell, NK-Cell, and Stroma-Derived Neoplasms of Lymphoid Tissues
Roberto N. Miranda, Catalina Amador, John K.C. Chan, Joan Guitart, Karen L. Rech, L. Jeffrey Medeiros, Kikkeri N. Naresh
Modern Pathology.2024; 37(8): 100512. CrossRef - Clinical epidemiology of Epstein-Barr virus-associated Lymphoproliferative Disorders (EBV-LPDs) in hospitalized children: A six-year multi-institutional study in China
Dilara Dilmurat, Xinyu Wang, Liwei Gao, Jiao Tian, Junhong Ai, Linlin Zhang, Mengjia Liu, Guoshuang Feng, Yueping Zeng, Ran Wang, Zhengde Xie
Italian Journal of Pediatrics.2024;[Epub] CrossRef - The fifth edition of the WHO‐Classification – what is new for cutaneous lymphomas?
Susanne Melchers, Jana D. Albrecht, Werner Kempf, Jan P. Nicolay
JDDG: Journal der Deutschen Dermatologischen Gesellschaft.2024; 22(9): 1254. CrossRef - An update on Epstein-Barr virus–and human T-lymphotropic virus type-1–induced cutaneous manifestations. CME Part II
Alejandro A. Gru, Jose A. Plaza, Jose A. Sanches, Denis Miyashiro, Omar P. Sangueza, Francisco Bravo Puccio, Sonia Toussaint, J. Martin Sangueza
Journal of the American Academy of Dermatology.2023; 88(5): 983. CrossRef - The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms
Rita Alaggio, Catalina Amador, Ioannis Anagnostopoulos, Ayoma D. Attygalle, Iguaracyra Barreto de Oliveira Araujo, Emilio Berti, Govind Bhagat, Anita Maria Borges, Daniel Boyer, Mariarita Calaminici, Amy Chadburn, John K. C. Chan, Wah Cheuk, Wee-Joo Chng,
Leukemia.2022; 36(7): 1720. CrossRef - Chronic active Epstein–Barr virus enteritis: A literature review
Yang Shen, Yu Fang Wang
Journal of Digestive Diseases.2022; 23(5-6): 248. CrossRef - EBV-Associated Lymphoproliferative Disorders
Young Hyeh Ko
Clinical Pediatric Hematology-Oncology.2021; 28(1): 14. CrossRef - Clinicopathologic findings of chronic active Epstein–Barr virus infection in adults: A single-center retrospective study in China
Jing Lin, Haicong Wu, Lei Gu, Xia Wu, Miaofang Su, Haiyan Lin, Bang Liu, Jiaolong Zheng, Xuan Mei, Dongliang Li
Clinical and Experimental Medicine.2021; 21(3): 369. CrossRef - Outcome of L-DEP regimen for treatment of pediatric chronic active Epstein–Barr virus infection
Honghao Ma, Liping Zhang, Ang Wei, Jun Yang, Dong Wang, Qing Zhang, Yunze Zhao, Sitong Chen, Hongyun Lian, Li Zhang, Chunju Zhou, Maoquan Qin, Zhigang Li, Tianyou Wang, Rui Zhang
Orphanet Journal of Rare Diseases.2021;[Epub] CrossRef - Epstein-Barr virus NK and T cell lymphoproliferative disease: report of a 2018 international meeting
Jeffrey I. Cohen, Keiji Iwatsuki, Young-Hyeh Ko, Hiroshi Kimura, Irini Manoli, Koichi Ohshima, Stefania Pittaluga, Leticia Quintanilla-Martinez, Elaine S. Jaffe
Leukemia & Lymphoma.2020; 61(4): 808. CrossRef - EBV-positive T/NK-associated lymphoproliferative disorders of childhood: A complete autopsy report
JonathanY Keow, WilliamM Stecho, AaronR Haig, NikhilA Sangle
Indian Journal of Pathology and Microbiology.2020; 63(1): 78. CrossRef - Chronic active Epstein‐Barr virus infection: A heterogeneous entity requiring a high index of suspicion for diagnosis
Sarah L. Ondrejka, Eric D. Hsi
International Journal of Laboratory Hematology.2020; 42(S1): 99. CrossRef - Epstein-Barr Virus-Associated T and NK-Cell Lymphoproliferative Diseases
Wook Youn Kim, Ivonne A. Montes-Mojarro, Falko Fend, Leticia Quintanilla-Martinez
Frontiers in Pediatrics.2019;[Epub] CrossRef - A clinicopathologic study of the spectrum of systemic forms of EBV‐associated T‐cell lymphoproliferative disorders of childhood: A single tertiary care pediatric institution experience in North America
Amy M. Coffey, Annisa Lewis, Andrea N. Marcogliese, M. Tarek Elghetany, Jyotinder N. Punia, Chung‐Che Chang, Carl E. Allen, Kenneth L. McClain, Amos S. Gaikwad, Nader Kim El‐Mallawany, Choladda V. Curry
Pediatric Blood & Cancer.2019;[Epub] CrossRef - Unusual lymphoid malignancy and treatment response in two children with Down syndrome
Ashley Geerlinks, Jennifer Keis, Bo Ngan, Amer Shammas, Reza Vali, Johann Hitzler
Pediatric Blood & Cancer.2019;[Epub] CrossRef - EBV-Positive Lymphoproliferations of B- T- and NK-Cell Derivation in Non-Immunocompromised Hosts
Stefan Dojcinov, Falko Fend, Leticia Quintanilla-Martinez
Pathogens.2018; 7(1): 28. CrossRef - Cutaneous Hematolymphoid and Histiocytic Proliferations in Children
Alejandro A Gru, Louis P Dehner
Pediatric and Developmental Pathology.2018; 21(2): 208. CrossRef - Clinicopathological categorization of Epstein–Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications
Jin Ho Paik, Ji-Young Choe, Hyojin Kim, Jeong-Ok Lee, Hyoung Jin Kang, Hee Young Shin, Dong Soon Lee, Dae Seog Heo, Chul-Woo Kim, Kwang-Hyun Cho, Tae Min Kim, Yoon Kyung Jeon
Leukemia & Lymphoma.2017; 58(1): 53. CrossRef - Cutaneous EBV-related lymphoproliferative disorders
Alejandro A. Gru, Elaine S. Jaffe
Seminars in Diagnostic Pathology.2017; 34(1): 60. CrossRef - T- and NK-Cell Lymphomas and Systemic Lymphoproliferative Disorders and the Immunodeficiency Setting
Dita Gratzinger, Daphne de Jong, Elaine S. Jaffe, Amy Chadburn, John K. C. Chan, John R. Goodlad, Jonathan Said, Yasodha Natkunam
American Journal of Clinical Pathology.2017; 147(2): 188. CrossRef - Systemic Epstein-Barr Virus-positive T-Cell Lymphoproliferative Disease of Childhood With Good Response to Steroid Therapy
Do-Hoon Kim, Myungshin Kim, Yonggoo Kim, Kyungja Han, Eunhee Han, Jae Wook Lee, Nack-Gyun Chung, Bin Cho
Journal of Pediatric Hematology/Oncology.2017; 39(8): e497. CrossRef - Recent advances in the risk factors, diagnosis and management of Epstein-Barr virus post-transplant lymphoproliferative disease
Paibel Aguayo-Hiraldo, Reuben Arasaratnam, Rayne H. Rouce
Boletín Médico del Hospital Infantil de México.2016; 73(1): 31. CrossRef - Severe Epstein–Barr virus infection in primary immunodeficiency and the normal host
Austen J. J. Worth, Charlotte J. Houldcroft, Claire Booth
British Journal of Haematology.2016; 175(4): 559. CrossRef - Recent advances in the risk factors, diagnosis and management of Epstein-Barr virus post-transplant lymphoproliferative disease
Paibel Aguayo-Hiraldo, Reuben Arasaratnam, Rayne H. Rouce
Boletín Médico Del Hospital Infantil de México (English Edition).2016; 73(1): 31. CrossRef - Epstein-Barr Virus–Associated Lymphomas
Ewelina Grywalska, Jacek Rolinski
Seminars in Oncology.2015; 42(2): 291. CrossRef - Epstein–Barr virus-associated T/natural killer-cell lymphoproliferative disorder in children and young adults has similar molecular signature to extranodal nasal natural killer/T-cell lymphoma but shows distinctive stem cell-like phenotype
Siok-Bian Ng, Koichi Ohshima, Viknesvaran Selvarajan, Gaofeng Huang, Shoa-Nian Choo, Hiroaki Miyoshi, Norio Shimizu, Renji Reghunathan, Hsin-Chieh Chua, Allen Eng-Juh Yeoh, Thuan-Chong Quah, Liang-Piu Koh, Poh-Lin Tan, Wee-Joo Chng
Leukemia & Lymphoma.2015; 56(8): 2408. CrossRef - An uncommon presentation of EBV-driven HLH. Primary or secondary? An ongoing dilemma
Tânia Serrão, Alexandra Dias, Pedro Nunes, António Figueiredo
BMJ Case Reports.2015; 2015: bcr2015209615. CrossRef - Hemophagocytic syndromes — An update
Gritta E. Janka, Kai Lehmberg
Blood Reviews.2014; 28(4): 135. CrossRef - Epstein–Barr virus‐associated T/natural killer‐cell lymphoproliferative disorders
Sanghui Park, Young H. Ko
The Journal of Dermatology.2014; 41(1): 29. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
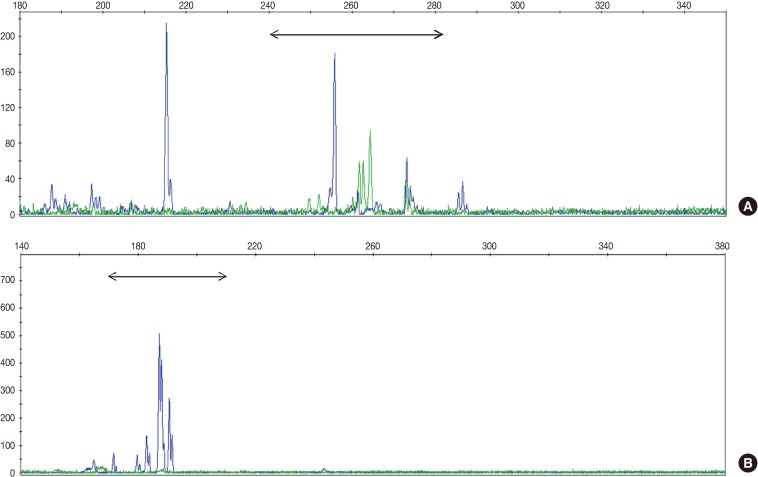
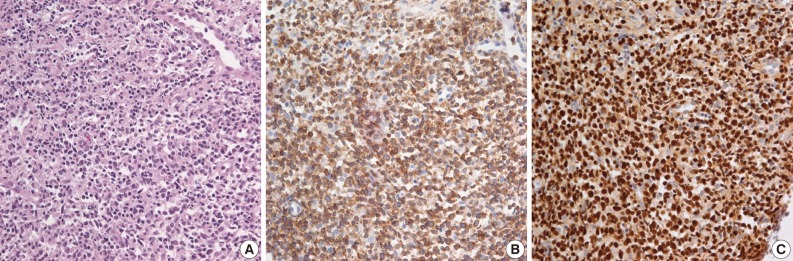
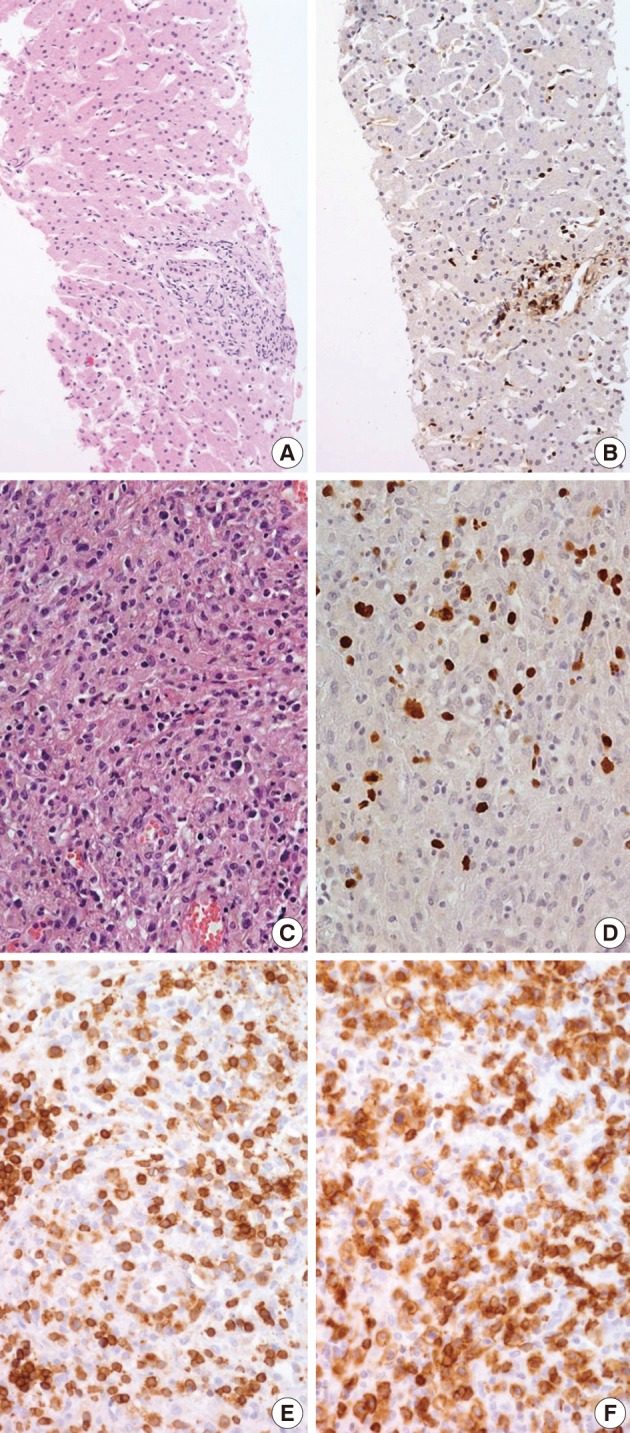
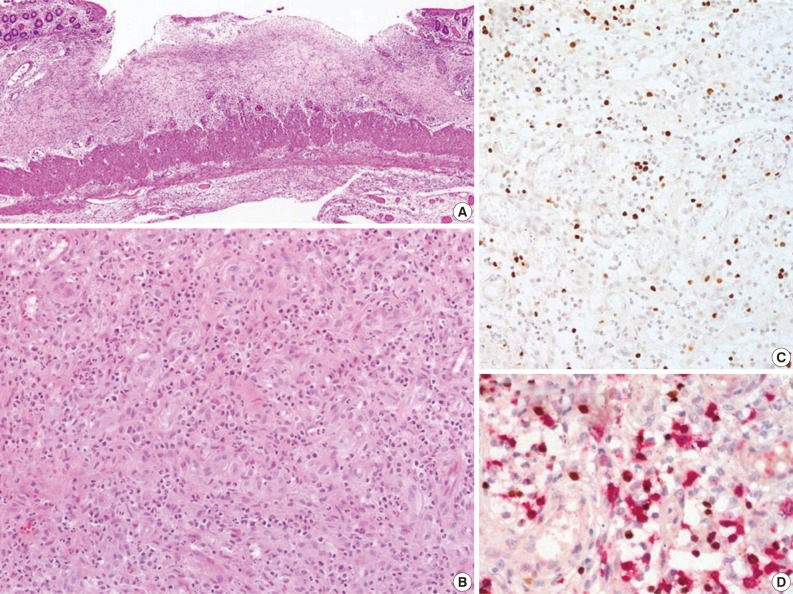




Fig. 1
Fig. 2
Fig. 3
Fig. 4
| No. | Sex | Age (yr) | Symptom | On-set | Biopsy site | Hemophagocytic histiocytosis | Cell size/Atypia | EBV-PCR (copy/μL whole blood) | EBV-ISH | EBV serology | IHC | TCRy gene | Treatment | Follow-up | Course |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Systemic T-cell LPD | |||||||||||||||
| 1 | F | 7 | Fever, cervical lymph node enlargement | 2MA | LN,BM | Present | Medium/moderate atypia | 42 (initial), 2.5-42 | Positive | EB-VCA, lgG(+) EB- | CD3+ | Monoclonal | VHR, L-Asp | 3 mo | Dead |
| VCA, IgM(-) | CD4<CD8 | ||||||||||||||
| EBV-EA(+) | CD56- | ||||||||||||||
| EBNA(+) | |||||||||||||||
| 2 | M | 43 | Fever, splenomegaly | 2MA | BM | Present | Small/mild atypia | 142 (initial), 0-142 | Positive | NC | CD3+ | Monoclonal | IMVP-16/PD | 2 mo | Dead |
| CD4+ | |||||||||||||||
| CD8+ | |||||||||||||||
| CD56– | |||||||||||||||
| EBV-positive hemophagocytic lymphohistiocytosis | |||||||||||||||
| 3 | F | 4 | Fever, jaundice, hepatomegaly, leukocytopenia | 2WA | LN, BM, liver | Present | Medium/severe atypia | 506 (initial), 4.77-6,422 | Positive | EB-VCA, igG(+) | CD3+ | Polyclonal | 106B, VHR | 3 mo | Dead |
| EB-VCA, lgM(+) | CD4- | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(-) | CD56– | ||||||||||||||
| 4 | M | 10 | Cervical lymph node enlargement, persistent fever, hepatosplenomegaly, LFT abnormality, pancytopenia, facial petechiae, gingival swelling | 1MA | BM | Present | Medium/moderate atypia | 5,810 (initial), 8.4-5, 810 | Failed | NA | CD3+ | No band | HLH-2004 |
69 mo | Alive |
| CD4- | |||||||||||||||
| CD8+ | |||||||||||||||
| CD56- | |||||||||||||||
| 5 | F | 20 | Headache, fever, night sweat, abdominal pain, hepatosplenomegaly | 3DA | BM | Present | Small/mild atypia | 8.5 | Positive | NA | CD3+ | Polyclonal | CHOP | 1 mo | Dead |
| CD4<CD8 | |||||||||||||||
| CD56– | |||||||||||||||
| 6 | M | 33 | Fever | 1MA | Liver | Present | Small/mild atypia | 105.7 | Positive | NA | CD3+ | Polyclonal | Steroid | 3 days | Dead |
| CD4+CD8+ | |||||||||||||||
| CD56- | |||||||||||||||
| 7 | F | 61 | Fever, chill | 4WA | Spleen, BM | Present | Small/mild atypia | NA | Positive | EB-VCA, igG(+) | CD3+ | Polyclonal | Steroid | 1 mo | Dead |
| EB-VCA, lgM(-) | CD4+ | ||||||||||||||
| EBV-EA(-) | CDS- | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| No. | Sex | Age (yr) | Symptom | On-set | Biopsy site | Hemophagocytic histiocytosis | Associated lymphoma | EBV-PCR (copy/μL whole blood) | EBV-ISH | EBV serology | IHC | TCRy gene | Treatment | Follow-up | Course |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 10 | LFT abnormality, hepatosplenomeg-aly, multiple enlarged lymph node (posterior neck, axilla and inguinal area) mosquito-bite hypersentivity | 2YA | LN, BM | Absent | Peripheral T-cell lymphoma | 77.6 (initial), 71.24-2,936 | Positive | EB-VCA, IgG(+) | CD3+ | Monoclonal/monomorphic | 106B | 10 mo | Dead |
| EB-VCA, IgM(–) | |||||||||||||||
| EBV-EA(+) | |||||||||||||||
| EBNA(+) | |||||||||||||||
| 2 | F | 14 | Fever, sore throat, IgA nephropathy, hydroa vacciniforme | Infancy | Skin, BM | Absent | T/NKcell lymphoma | NA | NA | NA | CD3+ | NA | CHOP, ESHAP | 18 mo | Dead |
| 3 | M | 15 | Nausea, weight loss, LFT abnormality, Herpes zoster infection | 2MA | Liver, BM | Present | None | 258.5 (initial), 22-258.5 | Positive | EB-VCA, IgG(+) | CD3+ | Monoclonal/monomorphic | HLH-2004 |
7 mo | Dead |
| EB-VCA, IgM(–) | CD4>CD8 | ||||||||||||||
| EBV-EA(±) | CD56– | ||||||||||||||
| EBNA(+) | |||||||||||||||
| 4 | M | 15 | Mosquito-bite hypersensitivity, palpable neck mass, NK lymphocytosis | 2WA | LN,BM | Absent | None | 529.8 (initial), 40-529.8 | Positive | EB-VCA, IgG(+) | CD3+ | NA | ABVD | 45 mo | Alive |
| EB-VCA, IgM(–) | CD4+ | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 5 | M | 16 | Fever and skin lesion (4YA), bowel perforation (7YA) mosquito-bite hypersensitivity, NK lymphocytosis | Infancy | Skin, BM | Present | None | 2,290 (initial), 7-2,290 | Positive | EB-VCA, IgG(+) | CD3+ | Polyclonal/polymorphic | HLH-2004 |
46 mo | Alive |
| EB-VCA, IgM(–) | CD4+ | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 6 | M | 21 | Mosquito-bite hypersensitivity, fever, epigastric pain, nausea, NK lymphocytosis | 7MA | Liver, BM | Present | None | NA | NA | EB-VCA, IgG(+) | NC | NA | IMVP-16PD | 3 mo | Dead |
| EB-VCA, IgM(–) | |||||||||||||||
| EBV-EA(±) | |||||||||||||||
| EBNA(+) | |||||||||||||||
| 7 | M | 21 | Chorea movement, hepatosplenomegaly, severe oral ulcer, history of pneumonia, thrombocytopenia, NK lymphocytosis | 2YA | Liver, BM | Present | None | 29 (initial), 29-73.5 | Positive | EB-VCA, IgG(+) | CD3+ | Polyclonal (KIR: NK cells with clonality) | CHOP | 10 mo | Alive (improved) |
| EB-VCA, IgM(–) | CD4>CD8 | ICE | |||||||||||||
| EBV-EA(–) | CD56+ | ||||||||||||||
| EBNA(+) | |||||||||||||||
| 8 | F | 29 | Fever, dizziness, nausea, hepatosplenomegaly, pancytopenia, LFT abnormality, diffuse lung infiltration | 2WA | LN, lung, BM | Absent | None | 189.2 (initial), 189.2-1,897 | Positive | EB-VCA, IgG(+) | CD3+ | Monoclonal/polymorphic | Self-limited | 6 mo | Alive (persistent) |
| EB-VCA, IgM(±) | CD4+ | ||||||||||||||
| EBV-EA(±) | CD8– | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 9 | M | 33 | Fatigue, hepatosplenomegaly, NK lymphocytosis | 7MA | Liver, BM | Present | None | 3,446 (initial), 1,918.6-12,112 | Positive | NA | CD3+ | NA | Refuse | 8 mo | Dead |
| CD4<CD8 | |||||||||||||||
| CD56– | |||||||||||||||
| 10 | F | 41 | Fever, hepatomegaly, abdominal pain, cerebral infact | 2YA | BM | Present | Medium/ mild atypia | 1,231.8 (initial), 1,231.8-16,188 | Positive | NA | CD3+ | Polyclonal/polymorphic | HLH-94 |
30 mo | Dead |
| CD4– | CVP | ||||||||||||||
| CD8+ | |||||||||||||||
| CD56– | |||||||||||||||
| 11 | M | 44 | LFT abnormality, hepatosplenomegaly, palpable neck mass | 8MA | LN, liver | Absent | None | NA | Positive | EB-VCA, IgG(+) | CD3+ | NA | Refuse | 6 mo | Dead |
| EB-VCA, IgM(-) | CD4+ | ||||||||||||||
| EBV-EA(+) | CD8+ | ||||||||||||||
| EBNA(+) | CD56– | ||||||||||||||
| 12 | F | 59 | Fever, myalgia, NK lymphocytosis | 7MA | BM | Present | None | 65.28 (initial), 36.6-2,934 | Negative | NA | CD3+ | No band | CVP, CHOP, IMVP-16/PD, VP | 4 mo | Dead |
| CD4– | |||||||||||||||
| CD8+ | |||||||||||||||
| CD56+ |
Treatment regimen; VHR: prednisolone, cyclophosphamide, daunorubicin, vincristine, L-asparaginase, intrathecal methotrexate, L-Asp: L-asparaginase, IMVP-16/PD: ifosfamide, methotrexate, etoposide, prednisolone, 106B: prednisolone, cyclophosphamide, daunorubicin, vincristine, L-asparaginase, CHOP: cyclophosphamide, doxorubicin, vincristine, prednisolone. LPD, lymphoproliferative disease; EBV, Epstein-Barr virus; PCR, polymerase chain reaction; ISH, HLH-94/2004: dexamethasone, cyclosporinA, intravenous Ig.
Treatment regimen; 106B: prednisolone, cyclophosphamide, daunorubicin, vincristine, L-asparaginase, CHOP: cyclophosphamide, daunorubicin, vincristine, prednisolone, ESHAP: etoposide, methylprednisolone, high-dose cytarabine, cisplatin, ABVD: adriamycin, bleomycin, vinblastine, dacarbazine, IMVP-16/PD: ifosfamide, methotrexate, etoposide, prednisolone, ICE: ifosfamide, carboplatin, etoposide, CVP: cyclophosphamide, vincristine, prednisolone. CAEBV, chronic active Epstein-Barr virus; EBV, Epstein-Barr virus; PCR, polymerase chain reaction; ISH, HLH-94/2004: dexamethasone, cyclosporinA, intravenous Ig.

