Follicular Dendritic Cell Sarcoma of the Inflammatory Pseudotumor-like Variant Presenting as a Colonic Polyp
Article information
Abstract
Follicular dendritic cell (FDC) sarcoma is rare and is classified either as conventional type or inflammatory pseudotumor (IPT)-like variant. Extranodal presentation is uncommon and nearly all gastrointestinal FDC tumors are of the conventional type. IPT-like variant tumors occur almost exclusively in the liver and spleen and are consistently associated with Epstein-Barr virus (EBV). Here we report the case of a 78-year-old woman with an IPT-like FDC sarcoma presenting as a pedunculated colonic polyp. Histologically, scanty atypical ovoid to spindle cells were mixed with a background of florid lymphoplasmacytic infiltrate, which led to an initial misdiagnosis of pseudolymphoma. These atypical cells expressed CD21, CD23, CD35, and D2-40, and were positive for EBV by in situ hybridization, confirming the diagnosis. The patient was free of disease five months after polypectomy without adjuvant therapy. Although extremely rare, the differential diagnosis for colonic polyp should include FDC sarcoma to avoid an erroneous diagnosis. A review of the 24 cases of IPT-like FDC sarcoma reported in the literature reveal that this tumor occurs predominantly in females with a predilection for liver and spleen, and has a strong association with EBV.
Follicular dendritic cells (FDC) are antigen-presenting cells normally residing in the germinal centers of secondary lymphoid follicles. First described in 1986 by Monda et al.,1 FDC sarcoma is a rare neoplasm comprising spindled to ovoid cells showing morphological and immunophenotypic features of FDC admixed with scattered small lymphocytes, i.e., the classical or conventional histopathology.1,2 In the inflammatory pseudotumor (IPT)-like variant, which occurs exclusively in the liver or spleen, the neoplastic spindle cells are scarce and are dispersed within a prominent lymphoplasmacytic infiltrate.2,3 One half to two thirds of FDC sarcomas are nodal lesions, most commonly in the cervical and axillary nodes; extranodal FDC sarcoma is rare with only around 100 cases being reported.4,5 Extranodal tumors preferentially affect the pelvic, abdominal and head and neck regions, with a minority involving the gastrointestinal (GI) tract.5,6 Morphologically, all of the hepatic lesions that have been described have belonged to the IPT-like variant, while most of the extrahepatic lesions including GI tumors were of the conventional type.5,6
Cheuk et al.3 reported the largest series of IPT-like FDC sarcoma, with all 11 cases occurring in the abdominal cavity and with overrepresentation of female patients and hepatosplenic involvement.3 Furthermore, all tumors in the series were positive for Epstein-Barr virus (EBV) by in situ hybridization. Here we report an EBV-positive, IPT-like variant of FDC sarcoma presenting as a colonic polyp, a peculiar presentation that has never been reported previously.
CASE REPORT
A 78-year-old female presented with several months of abdominal discomfort and bloody stool. The patient also reported irregular bowel movements. There was no fever, body weight loss, night sweating or malaise. Physical examination showed no evidence of lymphadenopathy. Laboratory data showed mild anemia with hemoglobin at 10 g/dL and normal carcinoembryonic antigen level. Barium enema performed one month prior to endoscopy revealed an oval-shaped filling defect in the colon. Colonoscopic examination revealed a 3 cm pedunculated polyp in the transverse colon, 50 cm from the anal verge (Fig. 1). An endoscopic biopsy showed granulation tissue with inflammatory exudates. Two weeks later, the patient underwent polypectomy. The resected tumor, 3.9×1.9×1.7 cm, was polypoid with ulceration. The cut surface was solid and tan without necrosis or hemorrhage. An initial diagnosis of pseudolymphoma was made, however, subsequent pathological review with additional ancillary studies lead to a revised diagnosis of FDC sarcoma. Abdominal computerized tomography and chest X-ray were negative. The patient was free of disease five months after the polypectomy without adjuvant chemotherapy.
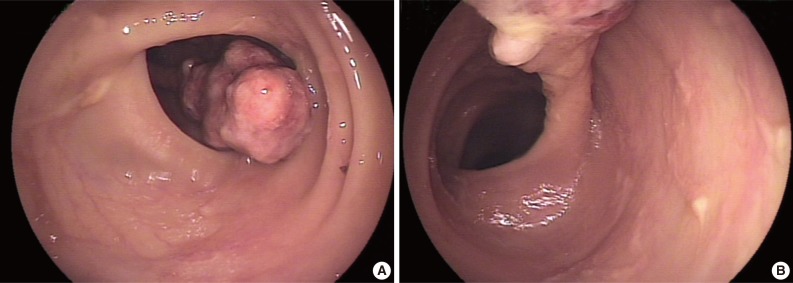
Endoscopy shows a pedunculated polyp in the colonic lumen (A) with a slender stalk (B).
Microscopically, the polypoid tumor showed focal ulceration and a florid lymphocytic infiltration with germinal center formation and a vascular stroma (Fig. 1). There were no epithelioid granulomas or hyalinized blood vessels entering the germinal centers. The interfollicular areas were extended by small lymphocytes and numerous plasma cells and included some large ovoid to spindled cells with vesicular nuclei and open chromatin with lightly eosinophilic syncytial cytoplasm. There were essentially no eosinophils visualized in the stroma. Occasionally, multinucleated giant cells and rare mitotic figures were discernible (Fig. 2). These atypical cells were dispersed and were more prominent beneath the area of ulceration. They were often accompanied by small sclerotic bands and were mostly scattered individually without forming intersecting spindle cell fascicles. Immunohistochemical studies showed that these large cells expressed CD21, CD23, CD35, CD45, p53, D2-40, and epithelial membrane antigen and weakly expressed estrogen receptors, but not CD34, CD68, CD117, DOG-1, S-100 protein, pan-cytokeratin, and progesterone receptors. Staining for smooth muscle actin was strongly positive in the spindle cells, but less intense in the areas where FDC markers such as CD21 were strongly expressed. The spindle cells were also positive for EBV-encoded mRNA (EBER) by in situ hybridization. The background lymphocytes were mixed small T and B cells; and the plasma cells were polytypic for light chain expression. The polymerase chain reaction-based clonality study for B-cell receptor gene rearrangement showed no amplification using IGH/FR3 primers. A diagnosis of IPT-like FDC sarcoma was made based on these results, while GI stromal tumor was excluded based on the histopathology and lack of staining for CD34, CD117, and DOG-1.
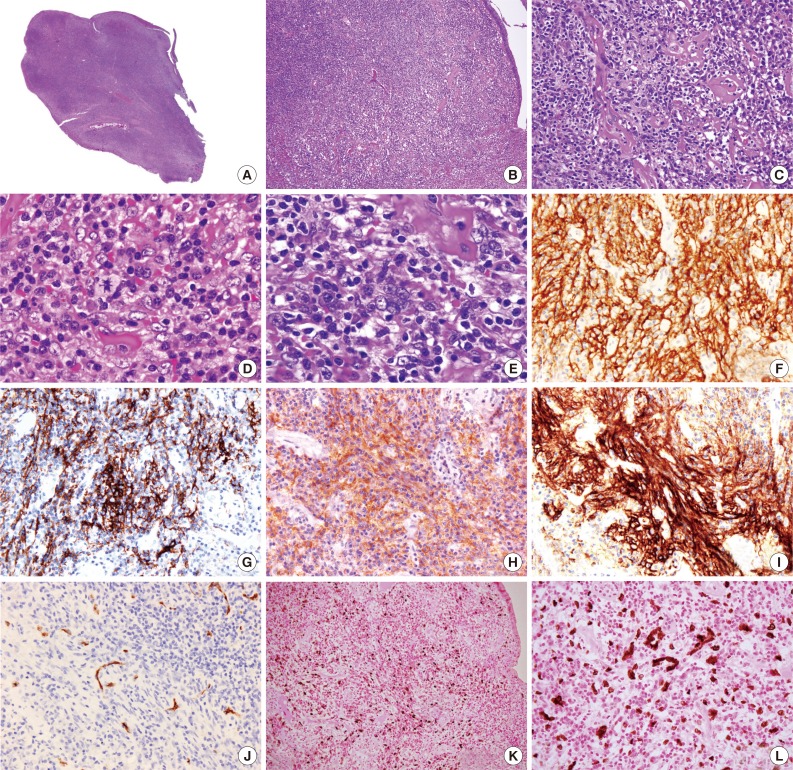
Photomicrography showing scanning power of the polyp with surface ulceration (A). (B, C) Low- and high-power views of polymorphic infiltration with scattered sclerotic bands beneath the colonic mucosa. (D, E) Oil immersion views reveal polygonal neoplastic cells with a mitosis (D, arrow) and multinucleation (E) in an inflammatory background with sclerosis. Immunohistochemical study shows that the tumor cells express CD21 (F), CD23 (G), CD35 (H), and D2-40 (I) but not CD34 (J). The tumor cells are positive for Epstein-Barr virus (K) with the high-power view highlighting the multinucleation (L) (in situ hybridization).
The pertinent clinicopathological features of cases with IPT-like FDC sarcoma in the English-language literature are summarized in Table 1. Overall there have been 20 women and 4 men (female:male ratio of 5:1) diagnosed with IPT-like FDC sarcoma, with a median age of 50 (range, 19 to 78). Excepting two cases, including the current one, all cases (92%) presented as hepatic or a splenic tumor. All the cases expressed at least one FDC marker with of the most frequent being CD21 (100%) and CAN.42 (100%), followed by CD23 (72%) and CD35 (67%). EBV-latent membrane protein 1 (LMP-1) was detected immunohistochemically in 75% of cases while all cases were positive for EBV by in situ hybridization. All cases, except the one described here, were treated with surgery and one case received additional adjuvant chemotherapy. Recurrence occurred in 4 (17%) patients with follow-up times ranging from 0.2 to 7.9 years. Only one patient (4%) died of disease at 7.9 years. In summary, IPT-like FDC sarcoma occurs predominantly in females, most commonly in the liver and spleen, displays a strong association with EBV, and generally has an indolent course.

Pertinent clinicopathological features of all IPT-like FDC sarcomas in the English-language literature
DISCUSSION
Extranodal FDC sarcoma remains under-recognized due to its rarity, and the diagnosis of the IPT-like variant is even more challenging. Although the polypoid presentation has never been reported previously in FDC sarcoma and the tumor cells were obscured by lymphoplasmacytic and stromal cells, some atypical cells with spindled or ovoid nuclei were still discernible in our case. Most importantly, the tumor cells could be highlighted using FDC markers (CD21, CD23, and CD35) and were diffusely positive for EBER. Our case demonstrated that when FDC sarcoma was included in the differential diagnostic list, a correct diagnosis could be reached even in a rare clinical setting using detailed morphological evaluation, immunohistochemistry and ancillary study with EBER.
Most FDC sarcomas present as lymphadenopathy, with the neck being the most common affected site.2 First reported by Chan et al.2 in 1994, extranodal FDC sarcomas are extremely rare with only around 100 cases in the English-language literature.4,5 They occur at various sites including the palate, tonsil, oral cavity, soft tissue, skin, mediastinum, liver, spleen, and GI tract. Extranodal FDC sarcomas involving the GI tract are extremely rare and the vast majority of these tumors are of the conventional type, circumscribed with yellowish white, fleshy cutting surfaces, ranging from 1 to 20 cm.17 Microscopically they comprise spindled to ovoid cells forming fascicles, a storiform growth pattern pattern, whorls, diffuse sheets or vague nodules. Scattered small lymphocytes are always present. The tumor cells show various degrees of nuclear atypia from benign to high-grade malignant features. Most commonly, the tumor cells form a syncytial pattern with indistinct cell borders containing oval or elongated nuclei and vesicular or granular finely dispersed chromatin and distinct nucleoli. Hemorrhage and necrosis are often present.18
The IPT-like variant of FDC sarcoma differs from the conventional type by a marked female predominance, a selective localization in the liver and spleen, frequent systemic symptoms, indolent behavior despite an intra-abdominal location and a dispersed distribution of tumor cells among prominent lymphoplasmacytic cells. Many of these tumors were initially reported in the literature as inflammatory pseudotumors. Most notably, this variant is consistently associated with EBV, in contrast to the conventional type, which is only very rarely associated with EBV.3 The universal association of EBV with IPT-like variant of FDC sarcomas is strongly suggestive of a pathogenetic role; EBV-induced cytokines and monokines are known to lead to vascular proliferation, inflammation and damage of the blood vessels,19 which may explain the unusual presentations of prominent vascular changes in our case.
All GI and IPT-like FDC sarcomas involving liver/spleen present as circumscribed fleshy masses with frequent hemorrhage and necrosis. Furthermore, previously reported GI tumors involved the intestinal wall or mesentery, and were nearly all of the conventional type. The exception is a single case of IPT-like variant reported by Agaimy and Wünsch6 in which the main tumor was identified in the mesentery (6 cm) of the terminal ileum with a submucosal spreading (1.5 cm).20 Interestingly, this tumor exhibited neoplastic cells with bizarre lobulated nuclei and prominent eosinophilia mimicking Hodgkin lymphoma. As in all the other previously reported GI tumors, this case was also EBER-negative.6 The tumor in our patient was unique as the first case of GI FDC sarcoma with a polypoid growth and an association with EBV.
A wide age range and equal sex distribution have been reported for FDC sarcoma, except for a marked female predilection in cases of the IPT-like variant. The clinical course is usually indolent, much like a low or intermediate-grade soft tissue sarcoma. Most patients are treated by complete surgical resection, with or without adjuvant radiotherapy or chemotherapy, though the local recurrence rate is more than 50% and metastases occur in about 25% of patients.2 In a series of 11 patients with IPT-like FDC sarcoma treated by surgical excision alone, the overall mortality rate was 11% and the recurrence rate was 33%.3 A recent review of 106 cases of extranodal FDC sarcomas showed that extranodal FDC tumors behave like soft tissue sarcomas and could be classified histologically into low- and high-grade lesions. Tumor recurrence was associated with tumors greater than or equal to 5 cm in size, having high-grade histology and a mitotic count greater than or equal to 5 per 10 high power field were associated with tumor recurrence.5 In the 18 cases of GI FDC sarcoma reviewed by Agaimy and Wunsch,6 33% of the patients developed recurrence in 1-84 months and 11% died of disease within a short mean follow-up time, suggesting that some patients might develop recurrence or metastasis if extended follow-up were obtained. Our patient received polypectomy alone without adjuvant therapy and was free of recurrence in 5 months. A longer follow-up is clearly necessary in this case.
The most important alternate diagnosis to consider in our patient is inflammatory fibroid polyp (IFP). Occurring most commonly in the terminal ileum, IFPs are rare mesenchymal tumors of the GI tract that consist of edematous spindle-shaped stromal cells and an inflammatory infiltrate rich in eosinophils.21 Pantanowitz et al.22 investigated 16 IFPs and found that stromal cells were diffusely positive for CD34 and fascin in all cases, and that these stromal cells were, in addition, immunoreactive for calponin and smooth muscle actin in 88% and 25% of cases, respectively. CD35 was also found to be focally reactive in the stromal cells.12 They were negative for CD21, CD23, and EBER. Based on these findings, they considered IFP to be a reactive lesion and suggested that the proliferating stromal cells were of dendritic cell origin, with some cases also exhibiting myofibroblastic features. In brief, IFP can be distinguished from IPT-like variant of FDC sarcoma by the abundance of eosinophils, CD34 immunoreactivity, and the absence of FDC markers other than CD35 (such as CD21 and CD23) and EBER-negativity. In our case, in addition to EBER-positivity, there were essentially no eosinophils and the tumor cells expressed all FDC markers including CD21, CD23, CD35, and D2-40. These results are more in keeping with FDC sarcoma than with IFP.
The other item on the differential diagnosis is IPT. Makhlouf and Sobin23 compared IPT (or inflammatory myofibroblastic tumors [IMFT]) and IFP of the GI tract. They found that in four of the 20 IPT/IMFT and five of 21 IFP, the spindle cell population were positive for EBV by in situ hybridization and immunohistochemistry for LMP-1.23 They considered IMFT synonymous with IPT and concluded that the lesions in the GI tract were extremely rare and differ clinically, histologically, and immunohistochemically from IFP. However, they did not address the significance of EBV in these lesions. In the current World Helath Organization (WHO) classification of GI tumors, IPT and IPT-like FDC sarcoma can be distinguished by the expression of FDC markers and EBER-positivity in the later tumors.21 The EBV-positive tumors described earlier in the literature might well be classified as IPT-like FDC sarcomas using the current WHO classification.
In summary, we presented a rare case of primary IPT-like FDC sarcoma in the GI tract. This tumor presented as a colonic polyp and was misdiagnosed initially as pseudolymphoma due to the heavy inflammatory background obscuring the scanty tumor cells. To avoid misdiagnosis, a high index of suspicion is warranted and FDC sarcoma should be considered in the differential diagnosis of a colonic polyp with a heavy inflammatory background on histological examination.
Notes
No potential conflict of interest relevant to this article was reported.