Placental Mesenchymal Dysplasia with Fetal Gastroschisis
Article information
Placental mesenchymal dysplasia (PMD) is a rare, benign condition characterized by placentomegaly and abnormal chorionic villi with vesicle formation, fibroblastic hyperplasia, and vascular abnormalities. This condition can be misdiagnosed as molar pregnancy due to similar ultrasonographic findings [1]; however, unlike molar pregnancy, a fetus is usually present and there is no risk of trophoblastic proliferative disease in PMD. The authors report a case of placental mesenchymal dysplasia at 15 weeks of gestation associated with fetal gastroschisis and high serum beta human chorionic gonadotropin (β-hCG) that mimicked molar pregnancy.
CASE REPORT
A 29-year-old female with abnormal ultrasonographic findings was transferred to our institution. The patient had a normal sequential screening with the exception of an elevated serum β-hCG of 165,870 mIU/mL. Serum α-fetoprotein was not evaluated. Transabdominal ultrasound at 14+0 weeks of gestation revealed a thick globular placenta measuring 6.5×4.5 cm with multiple small, hypoechoic areas. Subsequent ultrasound at 14+5 weeks of gestation demonstrated a large multicystic honeycomb-like placenta measuring 8.8×5.7 cm with no sign of blood flow within the lesion (Fig. 1A). A fetus was identified with a crown-rump length of 9.6 cm (Fig. 1B). Based on the clinical setting, partial hydatidiform mole was suspected. Therapeutic termination was performed at 15+0 weeks of gestation.
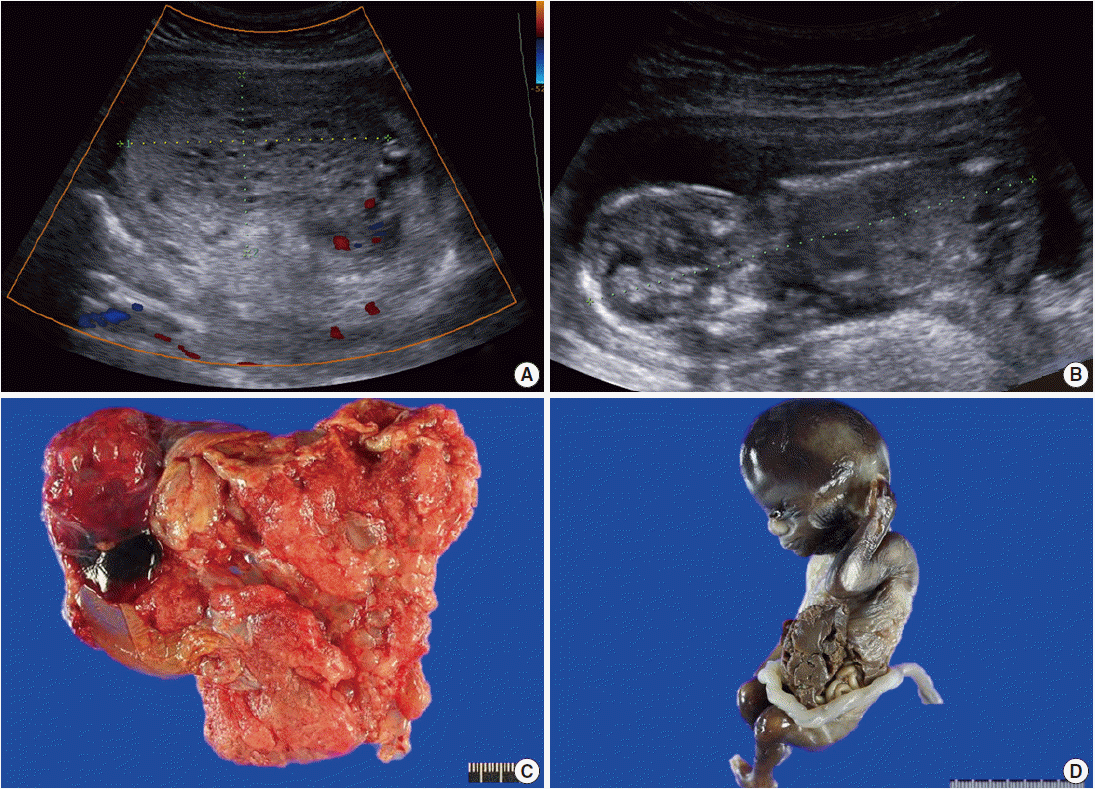
Radiologic and gross findings. (A) Transabdominal ultrasound at 14+5 weeks of gestation shows a large multicystic honeycomb-like placenta measuring 8.8×5.7 cm with no sign of blood flow within the lesion. (B) The fetus has a crown-rump length of 9.6 cm. (C) The placenta is markedly enlarged with multiple vesicles. (D) The fetus exhibits gastroschisis.
Grossly, the placenta was markedly enlarged with a diameter of 10 cm and the placental disc had multiple cystic vesicles; enlarged chorionic vessels were not found (Fig. 1C). A nonviable male fetus with a body weight of 73.57 g and with a crown-rump length of 9.8 cm, which were appropriate for gestational age, was delivered. However, he had a left lateral abdominal wall defect from which the liver and bowels protruded (Fig. 1D).
Microscopic examination of the placenta revealed several enlarged villi with cistern formation and abnormally thick vessels scattered among normal-sized terminal villi without abnormal trophoblast proliferation. Some chorionic villi showed proliferation of primitive stromal cells (Fig. 2A-C). Cytotrophoblasts were diffusely positive for p57KIP2 on immunohistochemical staining (Fig. 2D). Macroscopic and microscopic morphology confirmed a diagnosis of PMD accompanied by gastroschisis of the fetus.
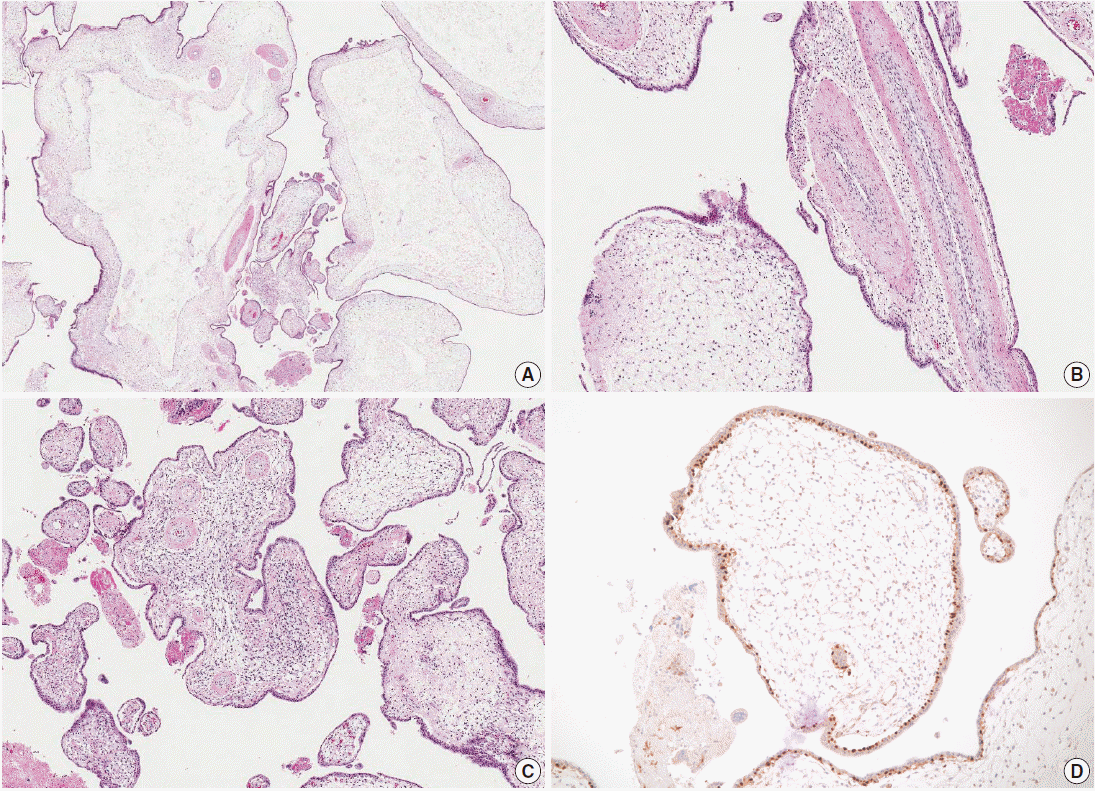
Microscopic findings of the placenta. (A) Cystic vesicles are surrounded by stromal tissue and trophoblasts, indicating cysts in enlarged stem villi. No abnormal trophoblast proliferation is observed. (B) The majority of the enlarged stem villi have abnormal vessels with thickened muscular walls. (C) Some of the villi show mesenchymal cell proliferation. (D) Cytotrophoblasts are diffusely immunopositive for p57KIP2.
DISCUSSION
PMD was first described as a rare, benign condition associated with an enlarged placenta in 1991 by Moscoso et al. [2]. The true incidence of PMD remains unclear, but it was reported to account for about 0.02% (7/30,758) of placentas examined at a Japanese hospital [3]. Most cases are asymptomatic and are diagnosed postpartum upon delivery of an abnormally large placenta. The typical sonographic feature of PMD is a thickened placenta with hypoechoic cystic spaces, which overlaps with that of molar pregnancy [4]. Thus, the differentiation of the two conditions may be difficult without placental pathology. One of the common laboratory abnormalities observed in PMD is increased maternal serum alpha fetoprotein, which is thought to originate from the fetus [2]. Serum β-hCG is typically normal to slightly elevated; the serum β-hCG level in this case was higher than that of normal pregnancy, which was unusual.
Grossly, the placenta is markedly enlarged for gestational age and the placental disc has multiple grape-like vesicles from 0.3 to 2.5 cm in size, which resemble those of molar pregnancy. Enlarged chorionic vessels are usually found in third-trimester PMD placentas, not seen in hydatidiform mole. However, prominent chorionic vessels may not be identified grossly during the early stages, as in this case. Histologically, enlarged villi with cistern formation and abnormally thick vessels are scattered among normal-sized terminal villi. Some chorionic villi show mesenchymal cell hyperplasia. Neither abnormal trophoblast proliferation nor scalloping of the villous surface, common histologic findings of hydatidiform mole, is present. p57KIP2 immunopositivity of the cytotrophoblasts in our case was helpful for excluding the possibility of complete hydatidiform mole [4].
The precise etiology of PMD has not been established. One theory is that it results from a congenital malformation of the mesoderm. Hypoxia and hypoperfusion of unknown origin may be responsible for the pathologic placental changes. More recently, androgenetic/biparental mosaicism has been suggested to be the underlying cause of PMD [4].
Unlike molar pregnancy, PMD is not associated with gestational trophoblastic diseases, but does increase the risk of intrauterine fetal growth restriction and intrauterine fetal death. Approximately 15% to 25% of PMD cases are associated with Beck-with-Wiedemann syndrome [5-7]. PMD and Beckwith-Wiedemann syndrome represent a broad spectrum of phenotypes with common etiology [4,8]. In our case, PMD was associated with fetal gastroschisis, which has not been reported previously [6]. A recently-favored hypothesis for the pathogenesis of gastroschisis is folding failure of the embryonic lateral body due to defective mesoderm [9]. Mesodermal abnormality is suggested to be the cause of the association between PMD and gastroschisis in this case.
In summary, PMD is a rare but clinically important placental abnormality that can be confused with partial hydatidiform mole and can be associated with abnormal fetal conditions. Therefore, PMD should be considered in the differential diagnosis of multicystic placenta. Awareness of this entity based on its sonographic similarity to molar pregnancy, unique pathologic features, and association with fetal complications needs to be emphasized in prenatal monitoring for the avoidance of unnecessary terminations.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.