Neuropathologic features of central nervous system hemangioblastoma
Article information

Abstract
Hemangioblastoma is a benign, highly vascularized neoplasm of the central nervous system (CNS). This tumor is associated with loss of function of the VHL gene and demonstrates frequent occurrence in von Hippel-Lindau (VHL) disease. While this entity is designated CNS World Health Organization grade 1, due to its predilection for the cerebellum, brainstem, and spinal cord, it is still an important cause of morbidity and mortality in affected patients. Recognition and accurate diagnosis of hemangioblastoma is essential for the practice of surgical neuropathology. Other CNS neoplasms, including several tumors associated with VHL disease, may present as histologic mimics, making diagnosis challenging. We outline key clinical and radiologic features, pathophysiology, treatment modalities, and prognostic information for hemangioblastoma, and provide a thorough review of the gross, microscopic, immunophenotypic, and molecular features used to guide diagnosis.
Hemangioblastoma is a benign neoplasm of the central nervous system (CNS) which most commonly occurs in the cerebellum. This tumor is highly vascular and contains neoplastic stromal cells with typically vacuolar cytoplasm. It is associated with loss or inactivation of the VHL gene, with frequent occurrence in von Hippel-Lindau (VHL) disease [1]. This entity has historically been referred to as “capillary hemangioblastoma,” “Lindau tumor,” and “angioblastoma,” although such terminologies are not currently recommended. Hemangioblastoma has an excellent prognosis with complete excision and is assigned a CNS World Health Organization (WHO) grade of 1 in the World Health Organization classification of CNS tumors [2]. Diagnosis of hemangioblastoma relies most heavily on histologic and immunophenotypic features with pieces of clinical, radiologic, and molecular information lending additional support.
EPIDEMIOLOGY
Hemangioblastomas are uncommon in the general population, with an overall incidence of 0.141 per 100,000 person-years in the United States [3]. They account for less than 2% of all CNS neoplasms but comprise an estimated 11% of primary posterior fossa tumors [4-6]. Some studies suggest a slightly higher incidence in male than in female patients, while others report no significant sex predilection [3,7,8]. Approximately 70% of cases are believed to be sporadic with the remaining 30% representing VHL-associated familial cases [9,10]. An estimated 60%–80% of patients with VHL disease develop CNS hemangioblastoma during their lifetime [11,12]. The average patient age at presentation varies by genetic subgroup, with VHL-associated tumors presenting on average two decades earlier than sporadic tumors; the mean age of onset is approximately 29 years in VHL-associated hemangioblastomas compared to 47 years in sporadic cases [9]. A comparison of epidemiologic data for sporadic and hereditary hemangioblastomas is summarized in Table 1.
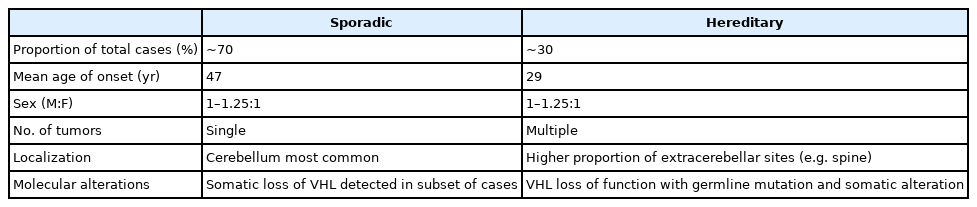
Summary of epidemiologic data for sporadic and hereditary hemangioblastomas
LOCALIZATION
Hemangioblastomas may arise throughout the CNS, most commonly involve the cerebellum, brainstem, and spinal cord, in order of descending frequency [3]. Less common sites of involvement include the supratentorial compartment and spinal nerve roots [13]. Cases of extraneural hemangioblastomas are rare [14,15]. While the cerebellum is still the most common site of hemangioblastoma in VHL patients, a higher proportion of extra-cerebellar sites are seen in VHL-associated cases, including retinal involvement in 28%–50% [16,17]. Patients with VHL disease are more likely to present with multiple hemangioblastomas [10,13,18]. Other VHL-associated neoplasms and their common sites of involvement include clear cell renal cell carcinoma (CCRCC) of the kidneys, paragangliomas of the head and neck, pheochromocytoma of the adrenal gland, neuroendocrine islet cell tumors of the pancreas, endolymphatic sac tumors of the inner ear, and cystadenomas of the epididymis and broad ligament [19].
ETIOPATHOGENESIS
Loss of function of the VHL tumor suppressor gene is implicated in most cases of hemangioblastoma, both familial and sporadic. VHL disease is an autosomal dominant tumor predisposition syndrome resulting from inactivating germline mutation in the VHL tumor suppressor gene [19,20]. This gene is located on the short arm of chromosome 3 (3p25-26) [21]. Biallelic inactivation of VHL is frequently found in familial cases of hemangioblastoma, according to Knudson’s “two-hit” hypothesis [22,23]. Under this system, a germline loss of function variant inactivates one copy of the VHL gene—the first hit—and loss of function of the remaining functional copy—the second hit—occurs prior to tumorigenesis [22]. The normally functioning VHL protein (pVHL) is involved in multiple cellular functions, including cell cycle regulation, apoptosis, and extracellular matrix formation [24-26]. pVHL also regulates cellular hypoxia signaling via the hypoxia-inducible factor (HIF) complex. Under normoxic conditions, the HIFα subunit undergoes hydroxylation creating a high affinity pVHL binding site [27,28]. As part of a multi-subunit ubiquitin ligase complex, pVHL facilitates HIFα polyubiquitylation and targeting for degradation [27-29]. In contrast, under hypoxic conditions, the HIF complex avoids hydroxylation and ubiquitin-associated degradation [30]. The stabilized complex serves as a transcription factor, upregulating the transcription of multiple growth factors to promote cell survival [31].
While hemangioblastoma pathogenesis is not yet fully understood, the pseudohypoxia hypothesis proposes a mechanism by which VHL loss promotes downstream tumorigenesis. Under this proposed pathway, VHL loss of function leads to loss of regulation of the HIF complex, which in turn promotes increased expression of growth factors in the absence of hypoxia, resulting in increased angiogenesis and conditions suitable for neoplastic growth [32].
HIF-independent pathways may also contribute to VHL disease and hemangioblastoma formation. VHL protein has been implicated in several other cellular functions, including regulation of apoptosis, microtubule stabilization, extracellular matrix formation, and cell-cell adhesion; loss of these functions could also contribute to tumorigenesis [33].
While the majority of familial cases demonstrate germline VHL loss, most studies indicate a smaller proportion of sporadic cases show VHL loss of function [34-36]. Historical studies of sporadic hemangioblastomas demonstrated VHL alterations in a minority of cases, although more recent investigation places the rate as high as 78% [36]. Older studies of sporadic hemangioblastoma may have shown erroneously low proportions of VHL loss due to low tumor purity. Since only the neoplastic stromal cells harbor the genetic alterations in sporadic cases, some alterations may evade detection when the stromal cells comprise a small fraction of the sampled tissue [36].
Epigenetic suppression may play a more prominent role in sporadic hemangioblastoma than in familial cases. One investigation found VHL promoter hypermethylation in 33% of sporadic hemangioblastomas but none of the VHL disease-associated cases [37]. Further studies are needed on sporadic cases without demonstrable VHL alterations to elucidate whether VHL loss is present but has not yet been detected, or if alternative pathways are responsible.
CLINICAL FEATURES
Clinical signs and symptoms of hemangioblastoma are mainly attributed to the tumor’s mass effect on adjacent regions, generalized increase in intracranial pressure, or obstruction of cerebrospinal fluid flow. General symptoms may include manifestations of increased intracranial pressure such as headaches, nausea, and emesis [10,38]. Secondary polycythemia may occur in approximately 5%–40% of cases, owing to ectopic production of erythropoietin [39,40].
Clinical presentation may vary widely based on anatomic localization of the tumor. Cerebellar tumors often present with dysmetria and ataxia [38]. In contrast, patients with spinal hemangioblastomas often present with symptoms associated with radiculopathy and myelopathy, including hypesthesia, weakness, hyperreflexia, pain, and incontinence [41,42]. Patients with brainstem hemangioblastomas may show evidence of cranial nerve impingement as well as increased intracranial pressure [43].
Some radiographic findings appear to correlate with symptomatology. Peritumoral cysts frequently underlie the clinical findings associated with hemangioblastomas, with at least 72% of symptomatic tumors but only 13% of asymptomatic tumors containing cysts [43]. In rare cases, hemangioblastomas may present with intraparenchymal, subarachnoid, or ventricular hemorrhage [44-46].
RADIOLOGIC FEATURES
Radiologic studies of hemangioblastoma generally show a welldemarcated enhancing mass ranging from solid to cystic, frequently presenting as a cyst with an enhancing mural nodule (Fig. 1) [47]. Tumors in the spinal cord are often associated with syrinx formation [48]. Computed tomography imaging demonstrates isodense signal compared to brain within the solid component [49]. Calcification is usually absent [49]. On magnetic resonance imaging, hemangioblastomas tend to appear as a T1 hypointense to isointense, T2 hyperintense nodule (Fig. 1) with serpentine flow voids in the nodular portion [47]. These tumors are often seen abutting the pia [50]. If a cystic component is present, the cyst wall rarely enhances [50].
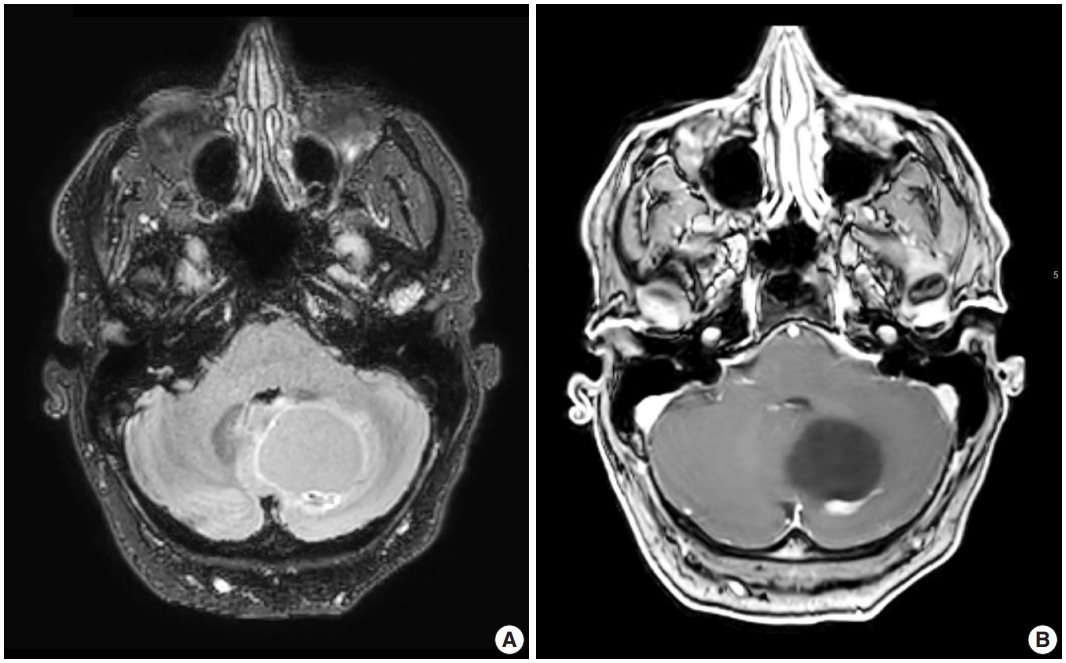
Representative brain magnetic resonance imaging for hemangioblastoma. (A) T2/FLAIR and (B) T1-weighted post-contrast sequences showing a posterior fossa cystic mass with a mural enhancing nodule.
MACROSCOPIC FEATURES
Grossly, hemangioblastomas appear as solid and/or cystic masses, often seen as a cyst with a mural nodule [43]. The tumors tend to be well-circumscribed and surrounded by a pseudocapsule [43]. Given the highly vascular nature of the neoplasm, cut surfaces may appear red in color, with yellow and orange variegation attributed to the high lipid content in neoplastic cells [51].
MICROSCOPIC FEATURES
Cytologic examination via direct smear or squash preparation can be a useful diagnostic aid at the time of intraoperative consultation. Unfortunately, hemangioblastoma samples are frequently resistant to squash preparation, resulting in thick clumps of tissue which may obfuscate cytologic detail (Fig. 2A) [52]. In the case of a successful cytologic smear preparation (or frozen section) with clearly visualized cells, the cytoplasm of neoplastic cells should demonstrate a characteristic foamy or finely vacuolated appearance (Fig. 2B) [52] . Cytoplasmic borders often appear indistinct, and nuclei may be mildly pleomorphic with hyperchromatic, speckled chromatin and occasional nuclear grooves [52]. Hemosiderin deposition is frequently seen, indicative of prior hemorrhage or red blood cell extravasation within these highly vascularized tumors [52].
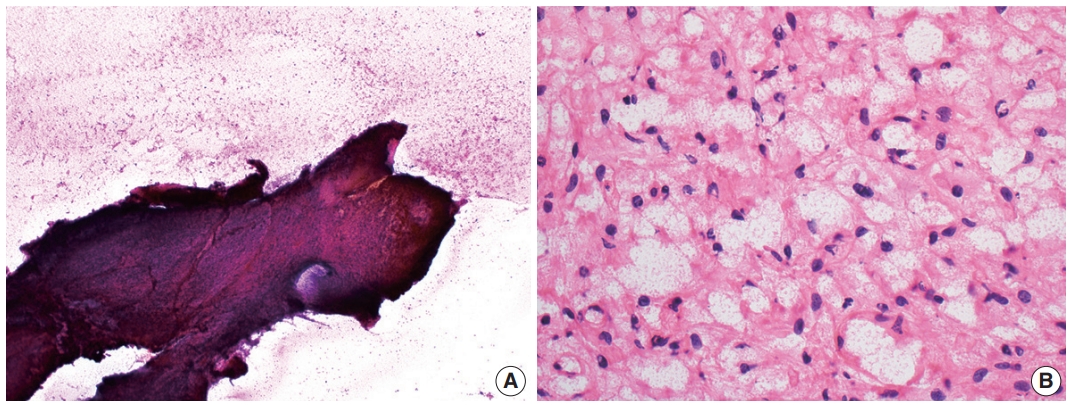
Intraoperative direct smear and frozen section findings of central nervous system hemangioblastoma. Representative intraoperative analysis of hemangioblastoma demonstrating (A) resistance to smear cytological preparation and (B) the presence of cells with vacuolated cytoplasm on frozen section.
On histologic sections, hemangioblastomas demonstrate compact, generally non-infiltrative growth patterns with variable lobularity [51]. The neoplastic component of these tumors are the stromal cells which are arranged within a rich network of thinwalled vessels (Fig. 3A) [51]. In these highly vascular tumors, there may be larger branching type vessels (Fig. 3B) and/or areas of hemorrhage (Fig. 3C). The neoplastic stromal cells classically contain clear, foamy cytoplasm with lipid-containing vacuoles (Fig. 3D) [51]. Nuclei may show mild pleomorphism, particularly in cases with degenerative atypia (Fig. 3E). Rare cytoplasmic hyaline globules may also be found [53,54].
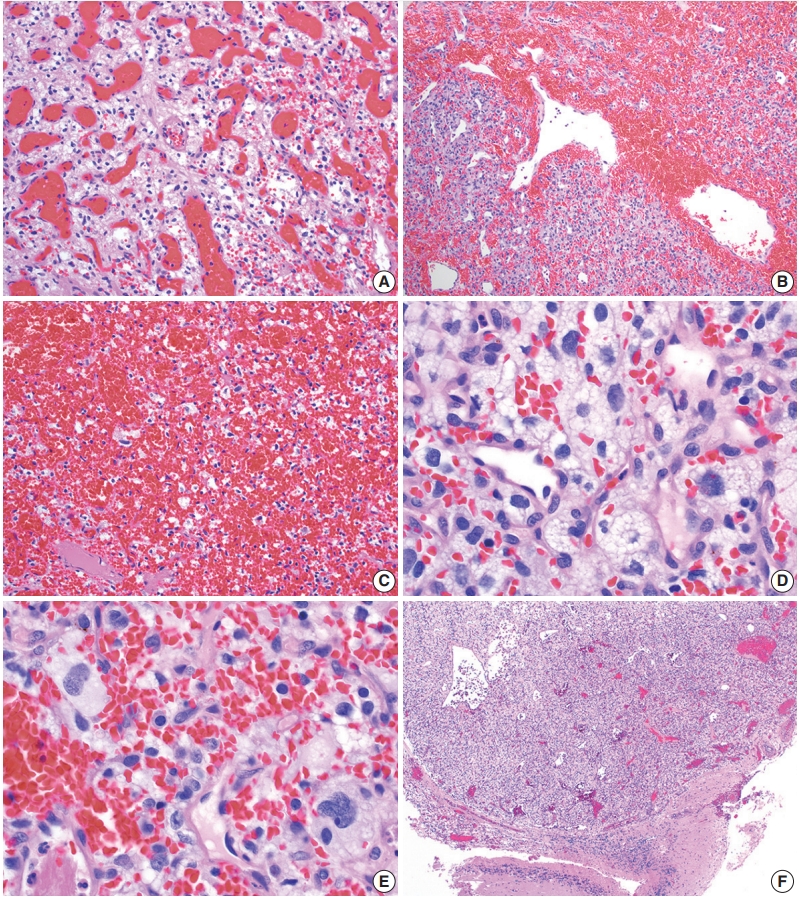
Histopathological features of central nervous system hemangioblastoma. Representative permanent sections highlighting how hemangioblastomas are highly vascular neoplasms and may contain (A) abundant capillaries, (B) large branching vessels, and/or (C) areas of hemorrhage. (D) Stromal cells contain abundant foamy vacuolated cytoplasm. (E) Nuclear atypia may be seen in some stromal cells. (F) Hemangioblastomas often from well-demarcated border with adjacent brain parenchyma.
Two histologic variants of hemangioblastoma have been described. The first is the more common reticular variant, characterized by a predominance of capillary vessels over stromal cells, and the second and less common variant is the cellular variant, marked by a preponderance of stromal cells and a less prominent vascular component [55].
If present, adjacent neural parenchyma maintains a well-demarcated border with the neoplasm (Fig. 3F), and may show marked reactive changes, including piloid gliosis with Rosenthal fibers [56]. Other helpful but non-essential features include the presence of intratumoral hemorrhage, mast cells, and cyst-like spaces. Extramedullary hematopoiesis may be found in approximately 15% of cases [57]. Mitotic figures are rare to absent.
IMMUNOHISTOCHEMICAL AND ANCILLARY STUDIES
Immunohistochemistry provides a useful and in some cases essential role in the diagnosis of hemangioblastoma and discrimination from histologic mimics.
Positive stains
Hemangioblastoma stromal cells classically show positive staining for inhibin alpha (Fig. 4A), NCAM1 (CD56), S100, and carbonic anhydrase IX & XII (Fig. 4B) [58-64]. Brachyury expression has been reported in 91% of cases and vimentin in 86–100% of cases [65,66]. Other positive stains include aquaporin 1, TFE3, and oil red O [61,62,67]. The background vascular component stains for endothelial cell markers such as CD31 (Fig. 4C) and CD34, while reticulin highlights vessel walls [62].
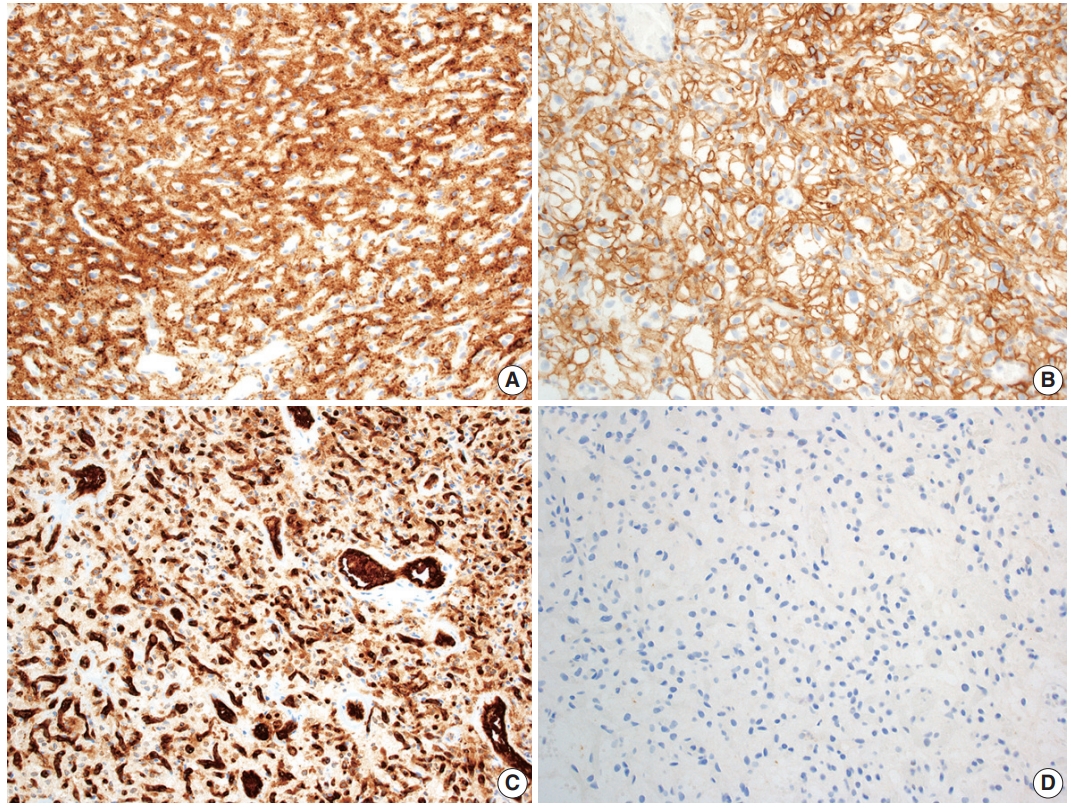
Immunohistochemical features of central nervous system hemangioblastoma. Immunohistochemical profiling for hemangioblastoma usually includes positivity for (A) inhibin alpha, (B) CAIX, and (C) CD31, with negativity for renal markers such as (D) PAX8.
Negative stains
By immunohistochemistry, the neoplastic cells are typically negative for AE1/AE3, CAM5.2, renal cell carcinoma (RCC) marker, and PAX8 (Fig. 4D) [58,60-62]. Other generally negative stains include PAX2 (positive in up to 5%), CD10 (positive in 0%–12%), epithelial membrane antigen (EMA; positive in 0%–36%), neuron-specific enolase (positive in 33%), and glial fibrillary acidic protein (GFAP; positive in scattered individual cells in up to 18%) [58,60,61,63,68].
Electron microscopy
Reports of electron microscopic studies of hemangioblastoma parallels histologic findings, with lipid-laden stromal cells occupying spaces between background capillaries [69].
Molecular and cytogenetic testing
Biallelic inactivation of the VHL tumor suppressor gene is present in many hemangioblastomas arising in VHL syndrome patients, with the majority of VHL-associated hemangioblastomas showing loss of heterozygosity of chromosome 3 [34,37]. VHL inactivation has been found in a large proportion of sporadic cases as well, with up to 78% of sporadic cases found to have loss or inactivation of VHL [34-37].
When a suspected hemangioblastoma shows ambiguous histopathologic and immunophenotypic features, newer ancillary studies such as DNA methylation testing and even microRNA (miRNA) analysis could aid diagnostic decision making. Studies of genome-wide DNA methylation array profiling report a distinctive epigenetic classifying signature for hemangioblastoma [69]. In addition, distinctive miRNA signatures have been described in hemangioblastomas, which show elevated miRNA-9 and decreased miRNA-200a expression compared to CCRCCs [70]. At present, these methodologies are not used in routine clinical practice, nor are they intended to replace histologic evaluation. However, there is a potential for more widespread use of these supplemental tests in the future as the technology becomes more accessible.
DIFFERENTIAL DIAGNOSIS
Several entities must be considered in the differential diagnosis of hemangioblastoma on a histopathologic basis. In the VHL disease patient, special care must be taken to exclude certain histologic mimics that are of greater risk due to genetic predisposition.
Clear cell renal cell carcinoma
One of the most common diagnostic challenges is differentiating hemangioblastoma from metastatic CCRCC, both due to the histologic overlap of vascular neoplasms with clear cytoplasm, as well as the increased risk of these entities in the setting of VHL syndrome. In contrast to the benign hemangioblastoma, CCRCC may demonstrate more noticeable cytologic atypia with prominent anaplasia and large nucleoli, although such features may be absent [60]. Other aggressive features that may be seen in CCRCC include mitotic activity and necrosis. Metastatic CCRCC cells typically show clear to eosinophilic cytoplasm but lack the foamy or vacuolated cytoplasm of hemangioblastoma stromal cells. By immunohistochemistry, CCRCC is usually positive for AE1/AE3, CAM5.2, EMA, CD10, PAX2, PAX8, and RCC marker, while negative for inhibin alpha, brachyury, and NCAM1 [59-62,65,71-73].
Meningioma
Certain histologic subtypes of meningioma may be mistaken for hemangioblastoma, particularly the clear cell, microcystic, and angiomatous variants which may demonstrate prominent vasculature and/or clear cytoplasm. In contrast to hemangioblastomas, meningiomas are typically positive for EMA in a weak, patchy, cytoplasmic pattern, along with SSTR2A in a membranous and cytoplasmic pattern [73,74]. Lower grade meningiomas, which would be more easily mistaken for a benign hemangioblastoma, often express nuclear progesterone receptor, although higher grade meningiomas are less likely to show expression [73]. Meningiomas should not express the hemangioblastoma markers inhibin alpha or brachyury [65,73].
Solitary fibrous tumor
Solitary fibrous tumor (SFT) is a fibroblastic tumor with variable histologic patterns. Like hemangioblastoma, it is a mesenchymal neoplasm with prominent vasculature. SFT may occur in a wide range of anatomic sites, although in neuropathologic practice, the intracranial dura is most often affected [75,76]. SFT is genetically defined by a NAB2 and STAT6 gene fusion due to inversion at the 12q13 locus [77-79]. This genomic alteration results in STAT6 nuclear immunoreactivity, which is a highly sensitive and specific marker [73,80,81]. SFT is negative for inhibin alpha [73].
Glial neoplasms
Glial neoplasms may occasionally need to be considered in the differential diagnosis of hemangioblastoma given the overlapping sites of disease within the CNS. Some glial neoplasms may also show circumscribed growth. While well preserved and well differentiated examples may be more easily distinguished from one another, pathologists may face greater challenges in the setting of small tissue samples, obscuring artifact, or poor differentiation. Cytologic smear preparations may be especially useful in the case of a well-differentiated glial neoplasm showing elongated glial fibrillary processes and poorly cohesive cells. However, caution must be exercised in interpretation of smears, as reactive astrocytes in gliotic neural parenchyma adjacent to a hemangioblastoma could show similar fibrillary glial processes. Histologic and immunophenotypic features vary more widely by glial tumor type.
Ependymomas generally show circumscribed growth, similar to hemangioblastomas, and may even show clear cell morphology in some cases [82]. In contrast, ependymomas may demonstrate characteristic perivascular pseudorosettes, ependymal rosettes, and ependymal canals. By immunohistochemistry, ependymomas show variable GFAP staining, often with accentuation of cell processes within perivascular pseudorosettes [83]. Of note, hemangioblastomas may also show rare GFAP immunoreactivity in a small number of cells [68]. While not an entirely specific feature, ependymomas may demonstrate paranuclear dot-like or ring-like staining with EMA [83].
Pilocytic astrocytomas, like hemangioblastomas, typically show circumscribed growth patterns. In contrast, pilocytic astrocytomas are characterized by a biphasic appearance, with compact fibrillar portions containing Rosenthal fibers and loose microcystic areas containing eosinophilic granular bodies. Pilocytic astrocytomas are generally positive for GFAP and Olig2 by immunohistochemistry and demonstrate characteristic mitogen-activated protein kinase pathway activating genetic alterations, of which KIAA1549-BRAF fusion is the most common [84-86].
Diffuse gliomas encompass a broad spectrum of low- and high-grade neoplasms, now organized into adult and pediatric types in the most recent iteration of the WHO classification of CNS tumors [2]. While the clinical presentation and genomic features of diffuse gliomas vary widely, they are characterized by individual neoplastic glioma cells infiltrating through background brain parenchyma. Entrapped neurons and axons may be detected by histology or by immunohistochemistry in regions with dense neoplastic growth. In contrast to hemangioblastomas, diffuse gliomas are generally positive for GFAP and Olig2 [85,87].
Paraganglioma/pheochromocytoma/cauda equina neuroendocrine tumor
Paragangliomas are circumscribed neuroendocrine neoplasms derived from the paraganglia of the sympathetic and parasympathetic nervous systems classically characterized by nested to organoid architecture of chief cells surrounded by sustentacular cells. Pheochromocytoma is the histologic correlate arising in the adrenal gland. While similar appearing neuroendocrine neoplasms of the cauda equina region have long been designated as paragangliomas, recent data suggests that these cauda equina neuroendocrine neoplasms are biologically distinct from paragangliomas elsewhere in the body based on DNA methylation profiling as well as the lack of SDH mutations [88,89]. Therefore, this entity has been renamed “cauda equina neuroendocrine tumor” in the fifth edition of the WHO classification of CNS tumors [2].
Paragangliomas, pheochromocytomas and cauda equina neuroendocrine tumors may show overlapping cytomorphologic features with hemangioblastomas. All these tumors may contain cells with clear to vacuolated cytoplasm in a richly vascular background [90]. The potential for confusion between these two entities is compounded by the fact that VHL patients are at increased risk of hemangioblastomas, parasympathetic paragangliomas, and pheochromocytomas. The organoid, “zellballen” architecture of paragangliomas, as well as their neuroendocrine nuclear features with uniform, round nuclei and salt and pepper chromatin may help to differentiate the entities, if these features are well preserved. On immunohistochemistry, paragangliomas and related entities are diffusely positive for synaptophysin and chromogranin, with intervening sustentacular cells staining for S-100 [90].
Hemangioma
Hemangioblastomas share some histologic features in common with hemangiomas, which are benign vascular neoplasms consisting of densely arranged capillaries and cavernous vessels. The tightly packed vessels contain plump endothelial cells which can impart the appearance of solid growth in areas. In the neuropathologic setting, hemangiomas are usually intraosseous, involving the spine and less frequently the skull, but have also been reported in CNS parenchyma [91-93]. If a suspected hemangioma involves the CNS parenchyma, adequate sampling and immunohistochemical workup should be undertaken to exclude the presence of neoplastic stromal cells.
TREATMENT AND PROGNOSIS
Hemangioblastoma is assigned a CNS WHO grade of 1, as complete excision is curative in most cases [2]. Surgical resection is considered the standard of care [94]. However, depending on the location of the tumor, excision may not be feasible. In the case of a symptomatic nonresectable tumor, stereotactic radiosurgery may provide some short-term control, but data on safety and long-term efficacy is currently limited [95-97].
Prospective natural history studies reveal unpredictable growth patterns in CNS hemangioblastoma. The predominant pattern is saltatory with intermittent periods of growth and quiescence [13,98]. Since it is difficult to predict the timeline of tumor growth, and because surgical intervention is not without risk of morbidity and mortality, treatment is often reserved for symptomatic tumors [94,98].
CNS hemangioblastoma carries a good prognosis with complete surgical excision, with significantly longer overall survival observed in patients who underwent gross total resection versus those who did not [96]. Compared to sporadic cases, outcomes are poorer in VHL patients; in this population, CNS hemangioblastomas remain the primary cause of mortality [99,100].
CONCLUSION
CNS hemangioblastoma is an important cause of morbidity and mortality in patients with VHL disease. Further studies are needed to clarify the pathogenesis of this disease, particularly in sporadic cases. Recognizing the basic clinical, radiographic, and pathologic features and considering a relevant differential diagnosis is crucial for making a correct diagnosis and guiding disease management.
Notes
Ethics Statement
Not applicable.
Availability of Data and Material
Data sharing not applicable to this article as no datasets were generated or analyzed during the study.
Code Availability
Not applicable.
Author Contributions
Conceptualization: RAY, PJC. Funding acquisition: PJC. Supervision: PJC. Writing—original draft: RAY, PJC. Writing—review & editing: RAY, PJC.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.
Acknowledgements
PJC is supported by the National Institutes of Health K08 CA245037 award.