 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 56(3); 2022 > Article
-
Review
Neuropathologic features of central nervous system hemangioblastoma -
Rebecca A. Yoda1,2
 , Patrick J. Cimino1
, Patrick J. Cimino1 -
Journal of Pathology and Translational Medicine 2022;56(3):115-125.
DOI: https://doi.org/10.4132/jptm.2022.04.13
Published online: May 3, 2022
1Department of Laboratory Medicine and Pathology, Division of Neuropathology, University of Washington, Seattle, WA, USA
2Department of Laboratory Medicine and Pathology, Division of Cytopathology, University of Washington, Seattle, WA, USA
- Corresponding Author: Rebecca A. Yoda, MD, Department of Laboratory Medicine and Pathology, University of Washington, 325 9th Avenue, Box 359791, Seattle, WA 98104-2499, USA Tel: +1-206-744-3145, Fax: +1-206-744-8240, E-mail: byoda@uw.edu
- This invited review is a featured collaboration with PathologyOutlines.com.
© 2022 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Hemangioblastoma is a benign, highly vascularized neoplasm of the central nervous system (CNS). This tumor is associated with loss of function of the VHL gene and demonstrates frequent occurrence in von Hippel-Lindau (VHL) disease. While this entity is designated CNS World Health Organization grade 1, due to its predilection for the cerebellum, brainstem, and spinal cord, it is still an important cause of morbidity and mortality in affected patients. Recognition and accurate diagnosis of hemangioblastoma is essential for the practice of surgical neuropathology. Other CNS neoplasms, including several tumors associated with VHL disease, may present as histologic mimics, making diagnosis challenging. We outline key clinical and radiologic features, pathophysiology, treatment modalities, and prognostic information for hemangioblastoma, and provide a thorough review of the gross, microscopic, immunophenotypic, and molecular features used to guide diagnosis.
- Hemangioblastomas are uncommon in the general population, with an overall incidence of 0.141 per 100,000 person-years in the United States [3]. They account for less than 2% of all CNS neoplasms but comprise an estimated 11% of primary posterior fossa tumors [4-6]. Some studies suggest a slightly higher incidence in male than in female patients, while others report no significant sex predilection [3,7,8]. Approximately 70% of cases are believed to be sporadic with the remaining 30% representing VHL-associated familial cases [9,10]. An estimated 60%–80% of patients with VHL disease develop CNS hemangioblastoma during their lifetime [11,12]. The average patient age at presentation varies by genetic subgroup, with VHL-associated tumors presenting on average two decades earlier than sporadic tumors; the mean age of onset is approximately 29 years in VHL-associated hemangioblastomas compared to 47 years in sporadic cases [9]. A comparison of epidemiologic data for sporadic and hereditary hemangioblastomas is summarized in Table 1.
EPIDEMIOLOGY
- Hemangioblastomas may arise throughout the CNS, most commonly involve the cerebellum, brainstem, and spinal cord, in order of descending frequency [3]. Less common sites of involvement include the supratentorial compartment and spinal nerve roots [13]. Cases of extraneural hemangioblastomas are rare [14,15]. While the cerebellum is still the most common site of hemangioblastoma in VHL patients, a higher proportion of extra-cerebellar sites are seen in VHL-associated cases, including retinal involvement in 28%–50% [16,17]. Patients with VHL disease are more likely to present with multiple hemangioblastomas [10,13,18]. Other VHL-associated neoplasms and their common sites of involvement include clear cell renal cell carcinoma (CCRCC) of the kidneys, paragangliomas of the head and neck, pheochromocytoma of the adrenal gland, neuroendocrine islet cell tumors of the pancreas, endolymphatic sac tumors of the inner ear, and cystadenomas of the epididymis and broad ligament [19].
LOCALIZATION
- Loss of function of the VHL tumor suppressor gene is implicated in most cases of hemangioblastoma, both familial and sporadic. VHL disease is an autosomal dominant tumor predisposition syndrome resulting from inactivating germline mutation in the VHL tumor suppressor gene [19,20]. This gene is located on the short arm of chromosome 3 (3p25-26) [21]. Biallelic inactivation of VHL is frequently found in familial cases of hemangioblastoma, according to Knudson’s “two-hit” hypothesis [22,23]. Under this system, a germline loss of function variant inactivates one copy of the VHL gene—the first hit—and loss of function of the remaining functional copy—the second hit—occurs prior to tumorigenesis [22]. The normally functioning VHL protein (pVHL) is involved in multiple cellular functions, including cell cycle regulation, apoptosis, and extracellular matrix formation [24-26]. pVHL also regulates cellular hypoxia signaling via the hypoxia-inducible factor (HIF) complex. Under normoxic conditions, the HIFα subunit undergoes hydroxylation creating a high affinity pVHL binding site [27,28]. As part of a multi-subunit ubiquitin ligase complex, pVHL facilitates HIFα polyubiquitylation and targeting for degradation [27-29]. In contrast, under hypoxic conditions, the HIF complex avoids hydroxylation and ubiquitin-associated degradation [30]. The stabilized complex serves as a transcription factor, upregulating the transcription of multiple growth factors to promote cell survival [31].
- While hemangioblastoma pathogenesis is not yet fully understood, the pseudohypoxia hypothesis proposes a mechanism by which VHL loss promotes downstream tumorigenesis. Under this proposed pathway, VHL loss of function leads to loss of regulation of the HIF complex, which in turn promotes increased expression of growth factors in the absence of hypoxia, resulting in increased angiogenesis and conditions suitable for neoplastic growth [32].
- HIF-independent pathways may also contribute to VHL disease and hemangioblastoma formation. VHL protein has been implicated in several other cellular functions, including regulation of apoptosis, microtubule stabilization, extracellular matrix formation, and cell-cell adhesion; loss of these functions could also contribute to tumorigenesis [33].
- While the majority of familial cases demonstrate germline VHL loss, most studies indicate a smaller proportion of sporadic cases show VHL loss of function [34-36]. Historical studies of sporadic hemangioblastomas demonstrated VHL alterations in a minority of cases, although more recent investigation places the rate as high as 78% [36]. Older studies of sporadic hemangioblastoma may have shown erroneously low proportions of VHL loss due to low tumor purity. Since only the neoplastic stromal cells harbor the genetic alterations in sporadic cases, some alterations may evade detection when the stromal cells comprise a small fraction of the sampled tissue [36].
- Epigenetic suppression may play a more prominent role in sporadic hemangioblastoma than in familial cases. One investigation found VHL promoter hypermethylation in 33% of sporadic hemangioblastomas but none of the VHL disease-associated cases [37]. Further studies are needed on sporadic cases without demonstrable VHL alterations to elucidate whether VHL loss is present but has not yet been detected, or if alternative pathways are responsible.
ETIOPATHOGENESIS
- Clinical signs and symptoms of hemangioblastoma are mainly attributed to the tumor’s mass effect on adjacent regions, generalized increase in intracranial pressure, or obstruction of cerebrospinal fluid flow. General symptoms may include manifestations of increased intracranial pressure such as headaches, nausea, and emesis [10,38]. Secondary polycythemia may occur in approximately 5%–40% of cases, owing to ectopic production of erythropoietin [39,40].
- Clinical presentation may vary widely based on anatomic localization of the tumor. Cerebellar tumors often present with dysmetria and ataxia [38]. In contrast, patients with spinal hemangioblastomas often present with symptoms associated with radiculopathy and myelopathy, including hypesthesia, weakness, hyperreflexia, pain, and incontinence [41,42]. Patients with brainstem hemangioblastomas may show evidence of cranial nerve impingement as well as increased intracranial pressure [43].
- Some radiographic findings appear to correlate with symptomatology. Peritumoral cysts frequently underlie the clinical findings associated with hemangioblastomas, with at least 72% of symptomatic tumors but only 13% of asymptomatic tumors containing cysts [43]. In rare cases, hemangioblastomas may present with intraparenchymal, subarachnoid, or ventricular hemorrhage [44-46].
CLINICAL FEATURES
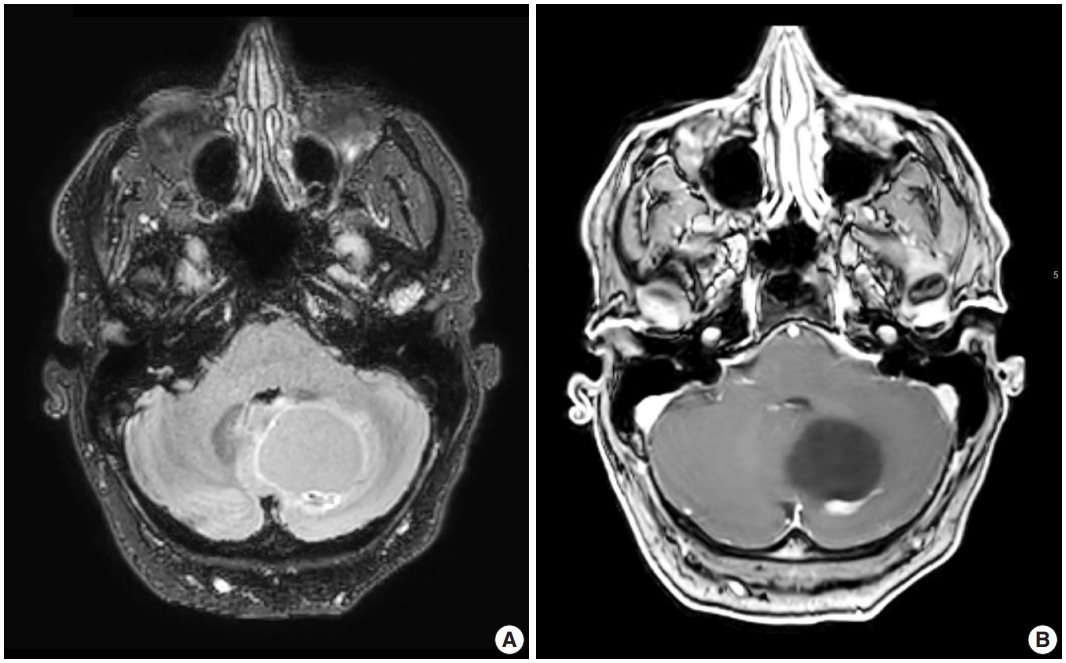
- Radiologic studies of hemangioblastoma generally show a welldemarcated enhancing mass ranging from solid to cystic, frequently presenting as a cyst with an enhancing mural nodule (Fig. 1) [47]. Tumors in the spinal cord are often associated with syrinx formation [48]. Computed tomography imaging demonstrates isodense signal compared to brain within the solid component [49]. Calcification is usually absent [49]. On magnetic resonance imaging, hemangioblastomas tend to appear as a T1 hypointense to isointense, T2 hyperintense nodule (Fig. 1) with serpentine flow voids in the nodular portion [47]. These tumors are often seen abutting the pia [50]. If a cystic component is present, the cyst wall rarely enhances [50].
RADIOLOGIC FEATURES
- Grossly, hemangioblastomas appear as solid and/or cystic masses, often seen as a cyst with a mural nodule [43]. The tumors tend to be well-circumscribed and surrounded by a pseudocapsule [43]. Given the highly vascular nature of the neoplasm, cut surfaces may appear red in color, with yellow and orange variegation attributed to the high lipid content in neoplastic cells [51].
MACROSCOPIC FEATURES
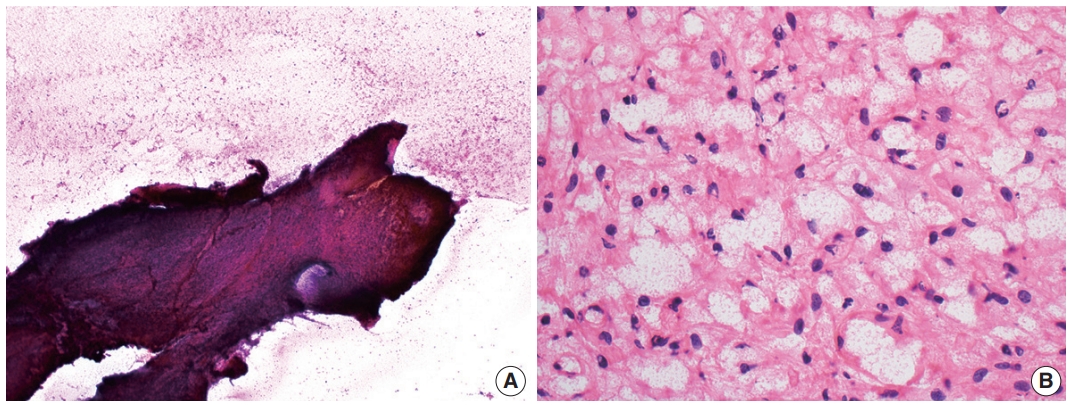
- Cytologic examination via direct smear or squash preparation can be a useful diagnostic aid at the time of intraoperative consultation. Unfortunately, hemangioblastoma samples are frequently resistant to squash preparation, resulting in thick clumps of tissue which may obfuscate cytologic detail (Fig. 2A) [52]. In the case of a successful cytologic smear preparation (or frozen section) with clearly visualized cells, the cytoplasm of neoplastic cells should demonstrate a characteristic foamy or finely vacuolated appearance (Fig. 2B) [52] . Cytoplasmic borders often appear indistinct, and nuclei may be mildly pleomorphic with hyperchromatic, speckled chromatin and occasional nuclear grooves [52]. Hemosiderin deposition is frequently seen, indicative of prior hemorrhage or red blood cell extravasation within these highly vascularized tumors [52].
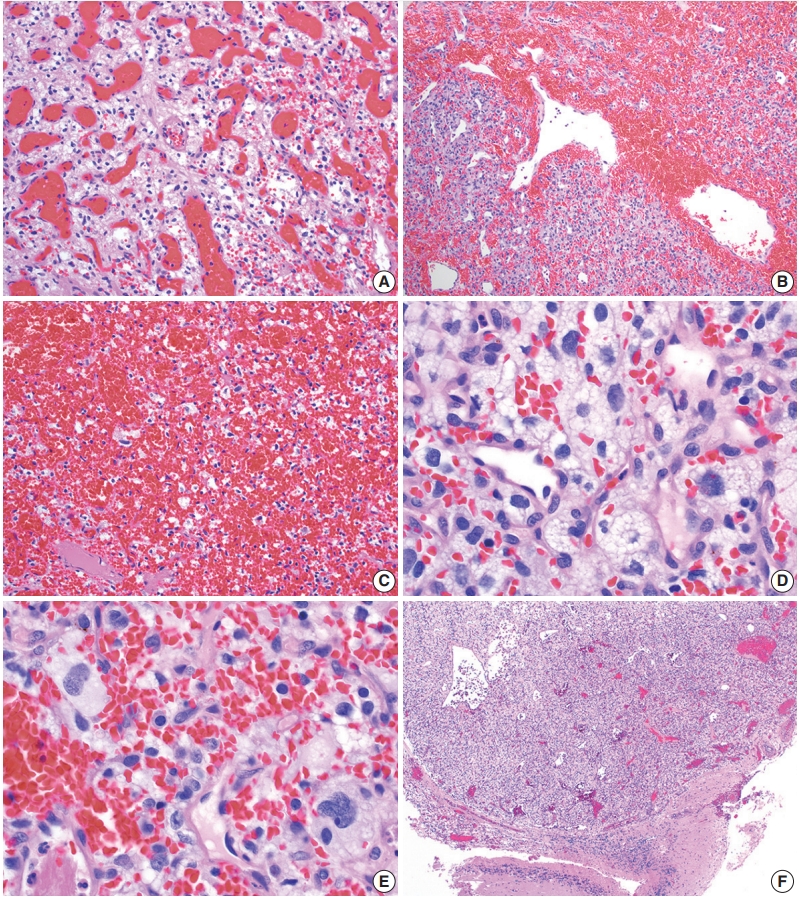
- On histologic sections, hemangioblastomas demonstrate compact, generally non-infiltrative growth patterns with variable lobularity [51]. The neoplastic component of these tumors are the stromal cells which are arranged within a rich network of thinwalled vessels (Fig. 3A) [51]. In these highly vascular tumors, there may be larger branching type vessels (Fig. 3B) and/or areas of hemorrhage (Fig. 3C). The neoplastic stromal cells classically contain clear, foamy cytoplasm with lipid-containing vacuoles (Fig. 3D) [51]. Nuclei may show mild pleomorphism, particularly in cases with degenerative atypia (Fig. 3E). Rare cytoplasmic hyaline globules may also be found [53,54].
- Two histologic variants of hemangioblastoma have been described. The first is the more common reticular variant, characterized by a predominance of capillary vessels over stromal cells, and the second and less common variant is the cellular variant, marked by a preponderance of stromal cells and a less prominent vascular component [55].
- If present, adjacent neural parenchyma maintains a well-demarcated border with the neoplasm (Fig. 3F), and may show marked reactive changes, including piloid gliosis with Rosenthal fibers [56]. Other helpful but non-essential features include the presence of intratumoral hemorrhage, mast cells, and cyst-like spaces. Extramedullary hematopoiesis may be found in approximately 15% of cases [57]. Mitotic figures are rare to absent.
MICROSCOPIC FEATURES
- Immunohistochemistry provides a useful and in some cases essential role in the diagnosis of hemangioblastoma and discrimination from histologic mimics.
- Positive stains
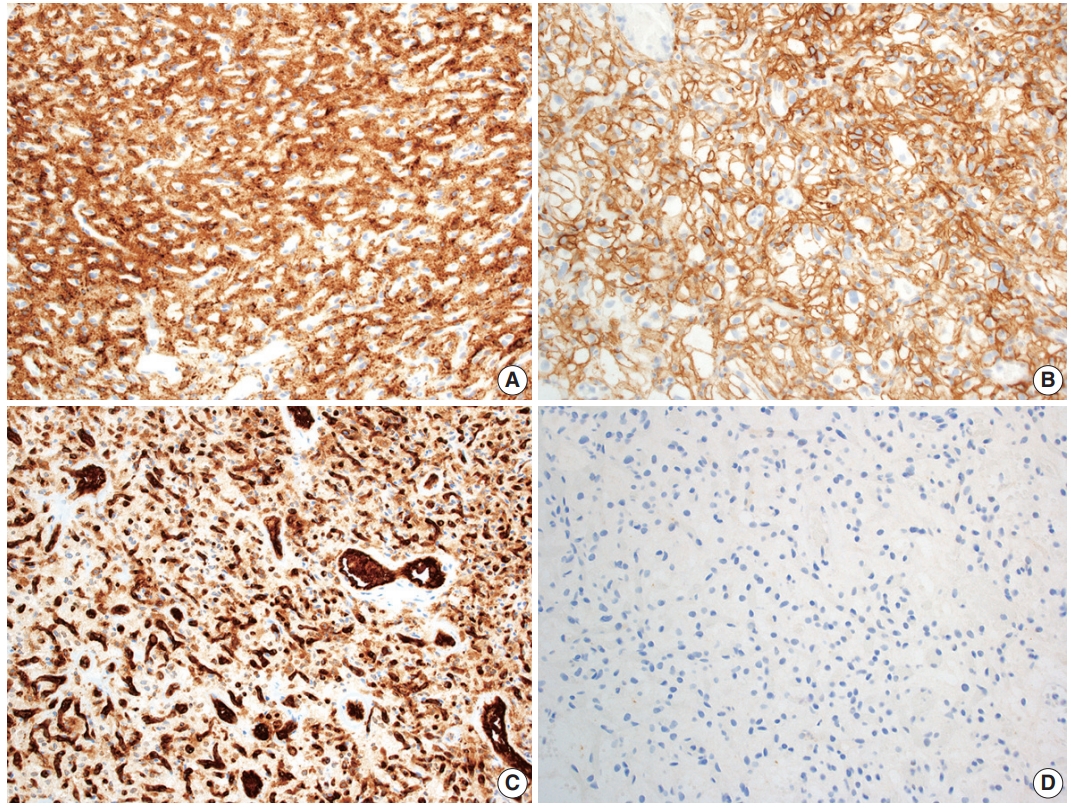
- Hemangioblastoma stromal cells classically show positive staining for inhibin alpha (Fig. 4A), NCAM1 (CD56), S100, and carbonic anhydrase IX & XII (Fig. 4B) [58-64]. Brachyury expression has been reported in 91% of cases and vimentin in 86–100% of cases [65,66]. Other positive stains include aquaporin 1, TFE3, and oil red O [61,62,67]. The background vascular component stains for endothelial cell markers such as CD31 (Fig. 4C) and CD34, while reticulin highlights vessel walls [62].
- Negative stains
- By immunohistochemistry, the neoplastic cells are typically negative for AE1/AE3, CAM5.2, renal cell carcinoma (RCC) marker, and PAX8 (Fig. 4D) [58,60-62]. Other generally negative stains include PAX2 (positive in up to 5%), CD10 (positive in 0%–12%), epithelial membrane antigen (EMA; positive in 0%–36%), neuron-specific enolase (positive in 33%), and glial fibrillary acidic protein (GFAP; positive in scattered individual cells in up to 18%) [58,60,61,63,68].
- Electron microscopy
- Reports of electron microscopic studies of hemangioblastoma parallels histologic findings, with lipid-laden stromal cells occupying spaces between background capillaries [69].
- Molecular and cytogenetic testing
- Biallelic inactivation of the VHL tumor suppressor gene is present in many hemangioblastomas arising in VHL syndrome patients, with the majority of VHL-associated hemangioblastomas showing loss of heterozygosity of chromosome 3 [34,37]. VHL inactivation has been found in a large proportion of sporadic cases as well, with up to 78% of sporadic cases found to have loss or inactivation of VHL [34-37].
- When a suspected hemangioblastoma shows ambiguous histopathologic and immunophenotypic features, newer ancillary studies such as DNA methylation testing and even microRNA (miRNA) analysis could aid diagnostic decision making. Studies of genome-wide DNA methylation array profiling report a distinctive epigenetic classifying signature for hemangioblastoma [69]. In addition, distinctive miRNA signatures have been described in hemangioblastomas, which show elevated miRNA-9 and decreased miRNA-200a expression compared to CCRCCs [70]. At present, these methodologies are not used in routine clinical practice, nor are they intended to replace histologic evaluation. However, there is a potential for more widespread use of these supplemental tests in the future as the technology becomes more accessible.
IMMUNOHISTOCHEMICAL AND ANCILLARY STUDIES
- Several entities must be considered in the differential diagnosis of hemangioblastoma on a histopathologic basis. In the VHL disease patient, special care must be taken to exclude certain histologic mimics that are of greater risk due to genetic predisposition.
- Clear cell renal cell carcinoma
- One of the most common diagnostic challenges is differentiating hemangioblastoma from metastatic CCRCC, both due to the histologic overlap of vascular neoplasms with clear cytoplasm, as well as the increased risk of these entities in the setting of VHL syndrome. In contrast to the benign hemangioblastoma, CCRCC may demonstrate more noticeable cytologic atypia with prominent anaplasia and large nucleoli, although such features may be absent [60]. Other aggressive features that may be seen in CCRCC include mitotic activity and necrosis. Metastatic CCRCC cells typically show clear to eosinophilic cytoplasm but lack the foamy or vacuolated cytoplasm of hemangioblastoma stromal cells. By immunohistochemistry, CCRCC is usually positive for AE1/AE3, CAM5.2, EMA, CD10, PAX2, PAX8, and RCC marker, while negative for inhibin alpha, brachyury, and NCAM1 [59-62,65,71-73].
- Meningioma
- Certain histologic subtypes of meningioma may be mistaken for hemangioblastoma, particularly the clear cell, microcystic, and angiomatous variants which may demonstrate prominent vasculature and/or clear cytoplasm. In contrast to hemangioblastomas, meningiomas are typically positive for EMA in a weak, patchy, cytoplasmic pattern, along with SSTR2A in a membranous and cytoplasmic pattern [73,74]. Lower grade meningiomas, which would be more easily mistaken for a benign hemangioblastoma, often express nuclear progesterone receptor, although higher grade meningiomas are less likely to show expression [73]. Meningiomas should not express the hemangioblastoma markers inhibin alpha or brachyury [65,73].
- Solitary fibrous tumor
- Solitary fibrous tumor (SFT) is a fibroblastic tumor with variable histologic patterns. Like hemangioblastoma, it is a mesenchymal neoplasm with prominent vasculature. SFT may occur in a wide range of anatomic sites, although in neuropathologic practice, the intracranial dura is most often affected [75,76]. SFT is genetically defined by a NAB2 and STAT6 gene fusion due to inversion at the 12q13 locus [77-79]. This genomic alteration results in STAT6 nuclear immunoreactivity, which is a highly sensitive and specific marker [73,80,81]. SFT is negative for inhibin alpha [73].
- Glial neoplasms
- Glial neoplasms may occasionally need to be considered in the differential diagnosis of hemangioblastoma given the overlapping sites of disease within the CNS. Some glial neoplasms may also show circumscribed growth. While well preserved and well differentiated examples may be more easily distinguished from one another, pathologists may face greater challenges in the setting of small tissue samples, obscuring artifact, or poor differentiation. Cytologic smear preparations may be especially useful in the case of a well-differentiated glial neoplasm showing elongated glial fibrillary processes and poorly cohesive cells. However, caution must be exercised in interpretation of smears, as reactive astrocytes in gliotic neural parenchyma adjacent to a hemangioblastoma could show similar fibrillary glial processes. Histologic and immunophenotypic features vary more widely by glial tumor type.
- Ependymomas generally show circumscribed growth, similar to hemangioblastomas, and may even show clear cell morphology in some cases [82]. In contrast, ependymomas may demonstrate characteristic perivascular pseudorosettes, ependymal rosettes, and ependymal canals. By immunohistochemistry, ependymomas show variable GFAP staining, often with accentuation of cell processes within perivascular pseudorosettes [83]. Of note, hemangioblastomas may also show rare GFAP immunoreactivity in a small number of cells [68]. While not an entirely specific feature, ependymomas may demonstrate paranuclear dot-like or ring-like staining with EMA [83].
- Pilocytic astrocytomas, like hemangioblastomas, typically show circumscribed growth patterns. In contrast, pilocytic astrocytomas are characterized by a biphasic appearance, with compact fibrillar portions containing Rosenthal fibers and loose microcystic areas containing eosinophilic granular bodies. Pilocytic astrocytomas are generally positive for GFAP and Olig2 by immunohistochemistry and demonstrate characteristic mitogen-activated protein kinase pathway activating genetic alterations, of which KIAA1549-BRAF fusion is the most common [84-86].
- Diffuse gliomas encompass a broad spectrum of low- and high-grade neoplasms, now organized into adult and pediatric types in the most recent iteration of the WHO classification of CNS tumors [2]. While the clinical presentation and genomic features of diffuse gliomas vary widely, they are characterized by individual neoplastic glioma cells infiltrating through background brain parenchyma. Entrapped neurons and axons may be detected by histology or by immunohistochemistry in regions with dense neoplastic growth. In contrast to hemangioblastomas, diffuse gliomas are generally positive for GFAP and Olig2 [85,87].
- Paraganglioma/pheochromocytoma/cauda equina neuroendocrine tumor
- Paragangliomas are circumscribed neuroendocrine neoplasms derived from the paraganglia of the sympathetic and parasympathetic nervous systems classically characterized by nested to organoid architecture of chief cells surrounded by sustentacular cells. Pheochromocytoma is the histologic correlate arising in the adrenal gland. While similar appearing neuroendocrine neoplasms of the cauda equina region have long been designated as paragangliomas, recent data suggests that these cauda equina neuroendocrine neoplasms are biologically distinct from paragangliomas elsewhere in the body based on DNA methylation profiling as well as the lack of SDH mutations [88,89]. Therefore, this entity has been renamed “cauda equina neuroendocrine tumor” in the fifth edition of the WHO classification of CNS tumors [2].
- Paragangliomas, pheochromocytomas and cauda equina neuroendocrine tumors may show overlapping cytomorphologic features with hemangioblastomas. All these tumors may contain cells with clear to vacuolated cytoplasm in a richly vascular background [90]. The potential for confusion between these two entities is compounded by the fact that VHL patients are at increased risk of hemangioblastomas, parasympathetic paragangliomas, and pheochromocytomas. The organoid, “zellballen” architecture of paragangliomas, as well as their neuroendocrine nuclear features with uniform, round nuclei and salt and pepper chromatin may help to differentiate the entities, if these features are well preserved. On immunohistochemistry, paragangliomas and related entities are diffusely positive for synaptophysin and chromogranin, with intervening sustentacular cells staining for S-100 [90].
- Hemangioma
- Hemangioblastomas share some histologic features in common with hemangiomas, which are benign vascular neoplasms consisting of densely arranged capillaries and cavernous vessels. The tightly packed vessels contain plump endothelial cells which can impart the appearance of solid growth in areas. In the neuropathologic setting, hemangiomas are usually intraosseous, involving the spine and less frequently the skull, but have also been reported in CNS parenchyma [91-93]. If a suspected hemangioma involves the CNS parenchyma, adequate sampling and immunohistochemical workup should be undertaken to exclude the presence of neoplastic stromal cells.
DIFFERENTIAL DIAGNOSIS
- Hemangioblastoma is assigned a CNS WHO grade of 1, as complete excision is curative in most cases [2]. Surgical resection is considered the standard of care [94]. However, depending on the location of the tumor, excision may not be feasible. In the case of a symptomatic nonresectable tumor, stereotactic radiosurgery may provide some short-term control, but data on safety and long-term efficacy is currently limited [95-97].
- Prospective natural history studies reveal unpredictable growth patterns in CNS hemangioblastoma. The predominant pattern is saltatory with intermittent periods of growth and quiescence [13,98]. Since it is difficult to predict the timeline of tumor growth, and because surgical intervention is not without risk of morbidity and mortality, treatment is often reserved for symptomatic tumors [94,98].
- CNS hemangioblastoma carries a good prognosis with complete surgical excision, with significantly longer overall survival observed in patients who underwent gross total resection versus those who did not [96]. Compared to sporadic cases, outcomes are poorer in VHL patients; in this population, CNS hemangioblastomas remain the primary cause of mortality [99,100].
TREATMENT AND PROGNOSIS
- CNS hemangioblastoma is an important cause of morbidity and mortality in patients with VHL disease. Further studies are needed to clarify the pathogenesis of this disease, particularly in sporadic cases. Recognizing the basic clinical, radiographic, and pathologic features and considering a relevant differential diagnosis is crucial for making a correct diagnosis and guiding disease management.
CONCLUSION
Ethics Statement
Not applicable.
Availability of Data and Material
Data sharing not applicable to this article as no datasets were generated or analyzed during the study.
Code Availability
Not applicable.
Author Contributions
Conceptualization: RAY, PJC. Funding acquisition: PJC. Supervision: PJC. Writing—original draft: RAY, PJC. Writing—review & editing: RAY, PJC.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.
Acknowledgments
- 1. Tse JY, Wong JH, Lo KW, Poon WS, Huang DP, Ng HK. Molecular genetic analysis of the von Hippel-Lindau disease tumor suppressor gene in familial and sporadic cerebellar hemangioblastomas. Am J Clin Pathol 1997; 107: 459-66. ArticlePubMed
- 2. WHO Classification of Tumours Editorial Board. Central nervous system tumours. 5th ed. Vol. 6 [Internet]. Lyon: International Agency for Research on Cancer, 2021 [cited 2022 Mar 10]. Available from: https://tumourclassification.iarc.who.int/chapters/45.
- 3. Yin X, Duan H, Yi Z, Li C, Lu R, Li L. Incidence, prognostic factors and survival for hemangioblastoma of the central nervous system: analysis based on the Surveillance, Epidemiology, and End Results Database. Front Oncol 2020; 10: 570103.ArticlePubMedPMC
- 4. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014-2018. Neuro Oncol 2021; 23: iii1-105. ArticlePubMedPMCPDF
- 5. Surawicz TS, McCarthy BJ, Kupelian V, Jukich PJ, Bruner JM, Davis FG. Descriptive epidemiology of primary brain and CNS tumors: results from the Central Brain Tumor Registry of the United States, 1990-1994. Neuro Oncol 1999; 1: 14-25. ArticlePubMedPMC
- 6. Constans JP, Meder F, Maiuri F, Donzelli R, Spaziante R, de Divitiis E. Posterior fossa hemangioblastomas. Surg Neurol 1986; 25: 269-75. ArticlePubMed
- 7. Westwick HJ, Giguere JF, Shamji MF. Incidence and prognosis of spinal hemangioblastoma: a Surveillance Epidemiology and End Results study. Neuroepidemiology 2016; 46: 14-23. ArticlePubMedPDF
- 8. Nguyen HS, Doan NB, Gelsomino M, et al. Intracranial hemangioblastoma: a SEER-based analysis 2004-2013. Oncotarget 2018; 9: 28009-15. ArticlePubMedPMC
- 9. Neumann HP, Eggert HR, Weigel K, Friedburg H, Wiestler OD, Schollmeyer P. Hemangioblastomas of the central nervous system: a 10-year study with special reference to von Hippel-Lindau syndrome. J Neurosurg 1989; 70: 24-30. PubMed
- 10. Conway JE, Chou D, Clatterbuck RE, Brem H, Long DM, Rigamonti D. Hemangioblastomas of the central nervous system in von Hippel-Lindau syndrome and sporadic disease. Neurosurgery 2001; 48: 55-62. ArticlePubMed
- 11. Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet 2003; 361: 2059-67. ArticlePubMed
- 12. Glasker S, Bender BU, Apel TW, et al. The impact of molecular genetic analysis of the VHL gene in patients with haemangioblastomas of the central nervous system. J Neurol Neurosurg Psychiatry 1999; 67: 758-62. ArticlePubMedPMC
- 13. Lonser RR, Butman JA, Huntoon K, et al. Prospective natural history study of central nervous system hemangioblastomas in von Hippel-Lindau disease. J Neurosurg 2014; 120: 1055-62. ArticlePubMedPMC
- 14. Doyle LA, Fletcher CD. Peripheral hemangioblastoma: clinicopathologic characterization in a series of 22 cases. Am J Surg Pathol 2014; 38: 119-27. PubMed
- 15. Nonaka D, Rodriguez J, Rosai J. Extraneural hemangioblastoma: a report of 5 cases. Am J Surg Pathol 2007; 31: 1545-51. PubMed
- 16. Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med 1990; 77: 1151-63. ArticlePubMed
- 17. Chew EY. Ocular manifestations of von Hippel-Lindau disease: clinical and genetic investigations. Trans Am Ophthalmol Soc 2005; 103: 495-511. PubMedPMC
- 18. Yousef A, Rutkowski MJ, Yalcin CE, Eren OC, Caliskan I, Tihan T. Sporadic and Von-Hippel Lindau disease-associated spinal hemangioblastomas: institutional experience on their similarities and differences. J Neurooncol 2019; 143: 547-52. ArticlePubMedPDF
- 19. Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet 2011; 19: 617-23. ArticlePubMedPMCPDF
- 20. Maher ER, Iselius L, Yates JR, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet 1991; 28: 443-7. ArticlePubMedPMC
- 21. Seizinger BR, Rouleau GA, Ozelius LJ, et al. Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature 1988; 332: 268-9. ArticlePubMedPDF
- 22. Knudson AG Jr, Strong LC, Anderson DE. Heredity and cancer in man. Prog Med Genet 1973; 9: 113-58. PubMed
- 23. Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol 2004; 22: 4991-5004. ArticlePubMed
- 24. Pause A, Lee S, Lonergan KM, Klausner RD. The von Hippel-Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. Proc Natl Acad Sci U S A 1998; 95: 993-8. ArticlePubMedPMC
- 25. Sendoel A, Kohler I, Fellmann C, Lowe SW, Hengartner MO. HIF-1 antagonizes p53-mediated apoptosis through a secreted neuronal tyrosinase. Nature 2010; 465: 577-83. ArticlePubMedPMCPDF
- 26. Tang N, Mack F, Haase VH, Simon MC, Johnson RS. pVHL function is essential for endothelial extracellular matrix deposition. Mol Cell Biol 2006; 26: 2519-30. ArticlePubMedPMCPDF
- 27. Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001; 292: 468-72. ArticlePubMed
- 28. Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001; 292: 464-8. ArticlePubMed
- 29. Yu F, White SB, Zhao Q, Lee FS. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A 2001; 98: 9630-5. PubMedPMC
- 30. Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med 2011; 365: 537-47. ArticlePubMed
- 31. Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011; 117: e207-17. ArticlePubMedPMCPDF
- 32. Favier J, Gimenez-Roqueplo AP. Pheochromocytomas: the (pseudo)-hypoxia hypothesis. Best Pract Res Clin Endocrinol Metab 2010; 24: 957-68. ArticlePubMed
- 33. Li M, Kim WY. Two sides to every story: the HIF-dependent and HIF-independent functions of pVHL. J Cell Mol Med 2011; 15: 187-95. ArticlePubMedPMC
- 34. Glasker S, Bender BU, Apel TW, et al. Reconsideration of biallelic inactivation of the VHL tumour suppressor gene in hemangioblastomas of the central nervous system. J Neurol Neurosurg Psychiatry 2001; 70: 644-8. ArticlePubMedPMC
- 35. Lee JY, Dong SM, Park WS, et al. Loss of heterozygosity and somatic mutations of the VHL tumor suppressor gene in sporadic cerebellar hemangioblastomas. Cancer Res 1998; 58: 504-8. PubMed
- 36. Shankar GM, Taylor-Weiner A, Lelic N, et al. Sporadic hemangioblastomas are characterized by cryptic VHL inactivation. Acta Neuropathol Commun 2014; 2: 167.ArticlePubMedPMCPDF
- 37. Takayanagi S, Mukasa A, Tanaka S, et al. Differences in genetic and epigenetic alterations between von Hippel-Lindau disease-related and sporadic hemangioblastomas of the central nervous system. Neuro Oncol 2017; 19: 1228-36. ArticlePubMedPMC
- 38. Jagannathan J, Lonser RR, Smith R, DeVroom HL, Oldfield EH. Surgical management of cerebellar hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg 2008; 108: 210-22. ArticlePubMed
- 39. Glasker S, Kruger MT, Klingler JH, et al. Hemangioblastomas and neurogenic polyglobulia. Neurosurgery 2013; 72: 930-5. ArticlePubMedPDF
- 40. Krieg M, Marti HH, Plate KH. Coexpression of erythropoietin and vascular endothelial growth factor in nervous system tumors associated with von Hippel-Lindau tumor suppressor gene loss of function. Blood 1998; 92: 3388-93. ArticlePubMedPDF
- 41. Roonprapunt C, Silvera VM, Setton A, Freed D, Epstein FJ, Jallo GI. Surgical management of isolated hemangioblastomas of the spinal cord. Neurosurgery 2001; 49: 321-7. ArticlePubMed
- 42. Chu BC, Terae S, Hida K, Furukawa M, Abe S, Miyasaka K. MR findings in spinal hemangioblastoma: correlation with symptoms and with angiographic and surgical findings. AJNR Am J Neuroradiol 2001; 22: 206-17. PubMedPMC
- 43. Wanebo JE, Lonser RR, Glenn GM, Oldfield EH. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg 2003; 98: 82-94. ArticlePubMed
- 44. Ene CI, Morton RP, Ferreira M Jr, Sekhar LN, Kim LJ. Spontaneous hemorrhage from central nervous system hemangioblastomas. World Neurosurg 2015; 83: 1180.Article
- 45. Glasker S, Van Velthoven V. Risk of hemorrhage in hemangioblastomas of the central nervous system. Neurosurgery 2005; 57: 71-6. ArticlePubMedPDF
- 46. de San Pedro JR, Rodriguez FA, Niguez BF, et al. Massive hemorrhage in hemangioblastomas literature review. Neurosurg Rev 2010; 33: 11-26. ArticlePubMedPDF
- 47. Ho VB, Smirniotopoulos JG, Murphy FM, Rushing EJ. Radiologic-pathologic correlation: hemangioblastoma. AJNR Am J Neuroradiol 1992; 13: 1343-52. PubMedPMC
- 48. Parizel PM, Baleriaux D, Rodesch G, et al. Gd-DTPA-enhanced MR imaging of spinal tumors. AJR Am J Roentgenol 1989; 152: 1087-96. ArticlePubMed
- 49. Ganti SR, Silver AJ, Hilal SK, Mawad ME, Sane P. Computed tomography of cerebellar hemangioblastomas. J Comput Assist Tomogr 1982; 6: 912-9. ArticlePubMed
- 50. Raz E, Zagzag D, Saba L, et al. Cyst with a mural nodule tumor of the brain. Cancer Imaging 2012; 12: 237-44. ArticlePubMedPMC
- 51. Ganeshan D, Menias CO, Pickhardt PJ, et al. Tumors in von Hippel-Lindau syndrome: from head to toe-comprehensive state-ofthe-art review. Radiographics 2018; 38: 849-66. ArticlePubMed
- 52. Commins DL, Hinton DR. Cytologic features of hemangioblastoma: comparison with meningioma, anaplastic astrocytoma and renal cell carcinoma. Acta Cytol 1998; 42: 1104-10. PubMed
- 53. Wang X, Haines GK 3rd, Mehrotra M, Houldsworth J, Si Q. Primary hemangioblastoma of the kidney with molecular analyses by next generation sequencing: a case report and review of the literature. Diagn Pathol 2022; 17: 34.ArticlePubMedPMCPDF
- 54. Shin Y, Kim S, Lee HW, Bang H, Suh YL. Supratentorial hemangioblastoma with unusual features. Korean J Pathol 2014; 48: 462-5. ArticlePubMedPMC
- 55. Hasselblatt M, Jeibmann A, Gerss J, et al. Cellular and reticular variants of haemangioblastoma revisited: a clinicopathologic study of 88 cases. Neuropathol Appl Neurobiol 2005; 31: 618-22. ArticlePubMed
- 56. Wippold FJ 2nd, Perry A, Lennerz J. Neuropathology for the neuroradiologist: rosenthal fibers. AJNR Am J Neuroradiol 2006; 27: 958-61. PubMedPMC
- 57. Zec N, Cera P, Towfighi J. Extramedullary hematopoiesis in cerebellar hemangioblastoma. Neurosurgery 1991; 29: 34-7. ArticlePubMed
- 58. Carney EM, Banerjee P, Ellis CL, et al. PAX2(-)/PAX8(-)/inhibin A(+) immunoprofile in hemangioblastoma: a helpful combination in the differential diagnosis with metastatic clear cell renal cell carcinoma to the central nervous system. Am J Surg Pathol 2011; 35: 262-7. PubMed
- 59. Hoang MP, Amirkhan RH. Inhibin alpha distinguishes hemangioblastoma from clear cell renal cell carcinoma. Am J Surg Pathol 2003; 27: 1152-6. ArticlePubMed
- 60. Rivera AL, Takei H, Zhai J, Shen SS, Ro JY, Powell SZ. Useful immunohistochemical markers in differentiating hemangioblastoma versus metastatic renal cell carcinoma. Neuropathology 2010; 30: 580-5. ArticlePubMed
- 61. Weinbreck N, Marie B, Bressenot A, et al. Immunohistochemical markers to distinguish between hemangioblastoma and metastatic clear-cell renal cell carcinoma in the brain: utility of aquaporin1 combined with cytokeratin AE1/AE3 immunostaining. Am J Surg Pathol 2008; 32: 1051-9. ArticlePubMed
- 62. Polydorides AD, Rosenblum MK, Edgar MA. Metastatic renal cell carcinoma to hemangioblastoma in von Hippel-Lindau disease. Arch Pathol Lab Med 2007; 131: 641-5. ArticlePubMedPDF
- 63. Hufnagel TJ, Kim JH, True LD, Manuelidis EE. Immunohistochemistry of capillary hemangioblastoma. Immunoperoxidase-labeled antibody staining resolves the differential diagnosis with metastatic renal cell carcinoma, but does not explain the histogenesis of the capillary hemangioblastoma. Am J Surg Pathol 1989; 13: 207-16. PubMed
- 64. Proescholdt MA, Mayer C, Kubitza M, et al. Expression of hypoxia-inducible carbonic anhydrases in brain tumors. Neuro Oncol 2005; 7: 465-75. ArticlePubMedPMC
- 65. Barresi V, Vitarelli E, Branca G, Antonelli M, Giangaspero F, Barresi G. Expression of brachyury in hemangioblastoma: potential use in differential diagnosis. Am J Surg Pathol 2012; 36: 1052-7. PubMed
- 66. Frank TS, Trojanowski JQ, Roberts SA, Brooks JJ. A detailed immunohistochemical analysis of cerebellar hemangioblastoma: an undifferentiated mesenchymal tumor. Mod Pathol 1989; 2: 638-51. PubMed
- 67. Yang Y, Gao H, Zhen T, et al. Hemangioblastoma: clinicopathologic study of 42 cases with emphasis on TFE3 expression. Am J Transl Res 2020; 12: 4498-510. PubMedPMC
- 68. Wizigmann-Voos S, Plate KH. Pathology, genetics and cell biology of hemangioblastomas. Histol Histopathol 1996; 11: 1049-61. PubMed
- 69. Capper D, Stichel D, Sahm F, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol 2018; 136: 181-210. ArticlePubMedPMCPDF
- 70. Venneti S, Boateng LA, Friedman JR, et al. MiRNA-9 and MiRNA-200a distinguish hemangioblastomas from metastatic clear cell renal cell carcinomas in the CNS. Brain Pathol 2012; 22: 522-9. ArticlePubMedPMC
- 71. Jung SM, Kuo TT. Immunoreactivity of CD10 and inhibin alpha in differentiating hemangioblastoma of central nervous system from metastatic clear cell renal cell carcinoma. Mod Pathol 2005; 18: 788-94. ArticlePubMedPDF
- 72. Sangoi AR, Karamchandani J, Kim J, Pai RK, McKenney JK. The use of immunohistochemistry in the diagnosis of metastatic clear cell renal cell carcinoma: a review of PAX-8, PAX-2, hKIM-1, RCCma, and CD10. Adv Anat Pathol 2010; 17: 377-93. PubMed
- 73. Boulagnon-Rombi C, Fleury C, Fichel C, Lefour S, Marchal Bressenot A, Gauchotte G. Immunohistochemical approach to the differential diagnosis of meningiomas and their mimics. J Neuropathol Exp Neurol 2017; 76: 289-98. ArticlePubMed
- 74. Menke JR, Raleigh DR, Gown AM, Thomas S, Perry A, Tihan T. Somatostatin receptor 2a is a more sensitive diagnostic marker of meningioma than epithelial membrane antigen. Acta Neuropathol 2015; 130: 441-3. ArticlePubMedPDF
- 75. Gold JS, Antonescu CR, Hajdu C, et al. Clinicopathologic correlates of solitary fibrous tumors. Cancer 2002; 94: 1057-68. ArticlePubMed
- 76. Mena H, Ribas JL, Pezeshkpour GH, Cowan DN, Parisi JE. Hemangiopericytoma of the central nervous system: a review of 94 cases. Hum Pathol 1991; 22: 84-91. ArticlePubMed
- 77. Chmielecki J, Crago AM, Rosenberg M, et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet 2013; 45: 131-2. ArticlePubMedPMCPDF
- 78. Schweizer L, Koelsche C, Sahm F, et al. Meningeal hemangiopericytoma and solitary fibrous tumors carry the NAB2-STAT6 fusion and can be diagnosed by nuclear expression of STAT6 protein. Acta Neuropathol 2013; 125: 651-8. ArticlePubMedPDF
- 79. Robinson DR, Wu YM, Kalyana-Sundaram S, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet 2013; 45: 180-5. ArticlePubMedPMCPDF
- 80. Doyle LA, Vivero M, Fletcher CD, Mertens F, Hornick JL. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol 2014; 27: 390-5. ArticlePubMedPDF
- 81. Yoshida A, Tsuta K, Ohno M, et al. STAT6 immunohistochemistry is helpful in the diagnosis of solitary fibrous tumors. Am J Surg Pathol 2014; 38: 552-9. ArticlePubMed
- 82. Fouladi M, Helton K, Dalton J, et al. Clear cell ependymoma: a clinicopathologic and radiographic analysis of 10 patients. Cancer 2003; 98: 2232-44. ArticlePubMed
- 83. Vege KD, Giannini C, Scheithauer BW. The immunophenotype of ependymomas. Appl Immunohistochem Mol Morphol 2000; 8: 25-31. ArticlePubMed
- 84. Tihan T, Ersen A, Qaddoumi I, et al. Pathologic characteristics of pediatric intracranial pilocytic astrocytomas and their impact on outcome in 3 countries: a multi-institutional study. Am J Surg Pathol 2012; 36: 43-55. PubMed
- 85. Ligon KL, Alberta JA, Kho AT, et al. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J Neuropathol Exp Neurol 2004; 63: 499-509. ArticlePubMed
- 86. Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013; 45: 602-12. ArticlePubMedPMCPDF
- 87. Nakagawa Y, Perentes E, Rubinstein LJ. Immunohistochemical characterization of oligodendrogliomas: an analysis of multiple markers. Acta Neuropathol 1986; 72: 15-22. ArticlePubMedPDF
- 88. Schweizer L, Thierfelder F, Thomas C, et al. Molecular characterization of CNS paragangliomas identifies cauda equina paragangliomas as a distinct tumor entity. Acta Neuropathol 2020; 140: 893-906. ArticlePubMedPMCPDF
- 89. Ramani B, Gupta R, Wu J, et al. The immunohistochemical, DNA methylation, and chromosomal copy number profile of cauda equina paraganglioma is distinct from extra-spinal paraganglioma. Acta Neuropathol 2020; 140: 907-17. ArticlePubMedPMCPDF
- 90. Koch CA, Mauro D, Walther MM, et al. Pheochromocytoma in von hippel-lindau disease: distinct histopathologic phenotype compared to pheochromocytoma in multiple endocrine neoplasia type 2. Endocr Pathol 2002; 13: 17-27. ArticlePubMed
- 91. Koga Y, Hamada S, Saito H, Akai T, Kuroda S. Intracranial, intraparenchymal capillary hemangioma: case report. NMC Case Rep J 2020; 7: 43-6. ArticlePubMedPMC
- 92. Fish C, Sy J, Wong J. High mitotic activity in a capillary hemangioma of the cauda equina: case report and review of the literature. Clin Neuropathol 2020; 39: 135-8. ArticlePubMed
- 93. Kasukurthi R, Ray WZ, Blackburn SL, Lusis EA, Santiago P. Intramedullary capillary hemangioma of the thoracic spine: case report and review of the literature. Rare Tumors 2009; 1: e10.ArticlePubMedPMCPDF
- 94. Dornbos D 3rd, Kim HJ, Butman JA, Lonser RR. Review of the Neurological Implications of von Hippel-Lindau Disease. JAMA Neurol 2018; 75: 620-7. ArticlePubMed
- 95. Kano H, Shuto T, Iwai Y, et al. Stereotactic radiosurgery for intracranial hemangioblastomas: a retrospective international outcome study. J Neurosurg 2015; 122: 1469-78. ArticlePubMed
- 96. Huang Y, Chan L, Bai HX, et al. Assessment of care pattern and outcome in hemangioblastoma. Sci Rep 2018; 8: 11144.ArticlePubMedPMCPDF
- 97. Asthagiri AR, Mehta GU, Zach L, et al. Prospective evaluation of radiosurgery for hemangioblastomas in von Hippel-Lindau disease. Neuro Oncol 2010; 12: 80-6. ArticlePubMedPMC
- 98. Ammerman JM, Lonser RR, Dambrosia J, Butman JA, Oldfield EH. Long-term natural history of hemangioblastomas in patients with von Hippel-Lindau disease: implications for treatment. J Neurosurg 2006; 105: 248-55. ArticlePubMed
- 99. Miyagami M, Katayama Y, Nakamura S. Clinicopathological study of vascular endothelial growth factor (VEGF), p53, and proliferative potential in familial von Hippel-Lindau disease and sporadic hemangioblastomas. Brain Tumor Pathol 2000; 17: 111-20. ArticlePubMedPDF
- 100. Binderup ML, Jensen AM, Budtz-Jorgensen E, Bisgaard ML. Survival and causes of death in patients with von Hippel-Lindau disease. J Med Genet 2017; 54: 11-8. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Hemangioblastoma of the Kidney—A Comprehensive Clinical, Pathological, and Genetic Analysis of Four Cases
Boglárka Pósfai, Alex Jenei, Gertrúd Forika, Attila Fintha, Zoltán Sápi, Áron Somorácz, Borbála Dénes, Ferenc Salamon, Kornélia Veronika Eizler, Nándor Giba, Dávid Semjén, Ildikó Illyés, Kristóf Attila Kovács, Gyöngyi Munkácsy, János Papp, Fanni Sánta, He
APMIS.2026;[Epub] CrossRef - Peritumoral Cystic Meningioma With Diagnostic Challenges: Two Case Reports and a Literature Review
Wei-Chih Chen, Zi-Jie Lin, Lam Chee-Tat
Cureus.2026;[Epub] CrossRef - Belzutifan-induced tumor regression in sporadic hemangioblastoma: a case report and literature review
Rebekka E. Hooks, Niket Yadav, Mark Willy L. Mondia, Georgios Mantziaris, Alaa Saleh, Anna Vi Jones, Matthew McCord, Melike Mut, Ashok R. Asthagiri, Benjamin W. Purow
Journal of Neuro-Oncology.2026;[Epub] CrossRef - Unusual immunohistochemical profiles in hemangioblastomas and their relevance in differential diagnosis: A comprehensive study of 112 cases
Jiri Soukup, Marie Novakova, Jan Hojny, Marketa Trnkova, Martin Syrucek, Tomas Jirasek, Patricie Delongova, Ales Kohout, Tomas Vebr, Jan Sroubek, Alena Sejkorova, Radim Lipina, Radim Brabec, Tomas Cesak, David Netuka
Journal of Neuropathology & Experimental Neurology.2026;[Epub] CrossRef - Immunohistochemical Expression of PAX8 in Central Nervous System Hemangioblastomas: A Potential Diagnostic Pitfall for Neuropathologists
Giuseppe Broggi, Jessica Farina, Valeria Barresi, Francesco Certo, Giuseppe Maria Vincenzo Barbagallo, Gaetano Magro, Rosario Caltabiano
Applied Immunohistochemistry & Molecular Morphology.2025; 33(3): 160. CrossRef - Endolymphatic Sac Tumor. Post-Radiosurgery Evaluation Using Time-Resolved Imaging of Contrast Kinetics MR Angiography
Antonella Blandino, Allegra Romano, Chiara Filippi, Sofia Pizzolante, Andrea Romano, Giulia Moltoni, Edoardo Covelli, Maurizio Barbara, Alessandro Bozzao
Ear, Nose & Throat Journal.2025;[Epub] CrossRef - Stereotactic radiosurgery in the management of central nervous system hemangioblastomas: a systematic review and meta-analysis
Amirhossein Zare, Amirhessam Zare, Alireza Soltani Khaboushan, Bardia Hajikarimloo, Jason P. Sheehan
Neurosurgical Review.2025;[Epub] CrossRef - Cerebellar medullary cistern hemangioblastoma
Dahai Cao, Qiang Zhang
Asian Journal of Surgery.2025; 48(9): 5843. CrossRef - Navigating rare vascular brain tumors: A retrospective observational study
Sana Ahuja, Dipanker S Mankotia, Naveen Kumar, Vyomika Teckchandani, Sufian Zaheer
Cancer Research, Statistics, and Treatment.2025; 8(2): 92. CrossRef - A potential new entity pending further validation of pulmonary primary interstitial Tumor: Lymphangioleiomyomatosis-like
Lingyu Zhao, Xiaochen Shen, Yun Niu, Huang Chen, Dingrong Zhong
Respiratory Medicine Case Reports.2025; 57: 102241. CrossRef - Renal cell carcinoma with fibromyomatous stroma (RCC FMS) and with hemangioblastoma‐like areas is part of the RCC FMS spectrum in patients with tuberous sclerosis complex
Katherina Baranova, Jacob A Houpt, Deaglan Arnold, Andrew A House, Laura Lockau, Lindsay Ninivirta, Stephen Pautler, Haiying Chen, Madeleine Moussa, Rola Saleeb, Jose A Gomez, Asli Yilmaz, Farshid Siadat, Adrian Box, Douglas J Mahoney, Franz J Zemp, Manal
Histopathology.2025; 87(5): 687. CrossRef - Renal hemangioblastoma and renal cell carcinoma with fibromyomatous stroma and hemangioblastoma-like areas belong to the spectrum of one entity
Kiril Trpkov, Norel Salut, Inmaculada Ribera-Cortada, Elías Tasso Xipell, Isabel Trias Puigsureda, Asli Yilmaz, Arjumand Riyaz Husain, Erik Nohr, Adrian Box, Farshid Siadat, Katherina Baranova, Rola M. Saleeb, Robert Stoehr, Arndt Hartmann, Abbas Agaimy
Virchows Archiv.2025;[Epub] CrossRef - Primary hemangioblastoma of rectum: a rare case report and review of literature
Aiping Zheng, Shaojuan Zhang, Qiang Ma, Wenxu Yang, Hualiang Xiao, Xinyu Liang
Journal of Cancer Research and Clinical Oncology.2025;[Epub] CrossRef - Cerebellar Hemangioblastoma Resection Complicated by Postoperative Vasogenic Edema in the Setting of Concurrent Immunotherapy Treatment
Aashka Sheth, Nicholas Dietz, Andrea Becerril-Gaitan, Rahim Kasem, Akshitkumar Mistry, Brian J Williams, Dale Ding, Isaac Abecassis
Cureus.2025;[Epub] CrossRef - Familial Von Hippel–Lindau Disease: A Case Series of Cerebral Hemangioblastomas with MRI, Histopathological, and Genetic Correlations
Claudiu Matei, Ioana Boeras, Dan Orga Dumitriu, Cosmin Mutu, Adriana Popescu, Mihai Gabriel Cucu, Alexandru Calotă-Dobrescu, Bogdan Fetica, Diter Atasie
Life.2025; 15(11): 1649. CrossRef - Characterization of spinal hemangioblastomas in patients with and without von Hippel-Lindau, and YAP expression
Ana-Laura Calderón-Garcidueñas, Steven-Andrés Piña-Ballantyne, Eunice-Jazmín Espinosa-Aguilar, Rebeca de Jesús Ramos-Sánchez
Revista Española de Patología.2024; 57(3): 160. CrossRef - Patients With Hemangioblastoma: Mood Disorders and Sleep Quality
Ali Riazi, Yaser Emaeillou, Nima Najafi, Mohammad Hoseinimanesh, Mohammad Ibrahim Ashkaran, Donya Sheibani Tehrani
Brain Tumor Research and Treatment.2024; 12(2): 87. CrossRef - Radiosurgically Treated Recurrent Cerebellar Hemangioblastoma: A Case Report and Literature Review
François Fabi, Ève Chamberland, Myreille D’Astous, Karine Michaud, Martin Côté, Isabelle Thibault
Current Oncology.2024; 31(7): 3968. CrossRef - Dual manifestations: spinal and cerebellar hemangioblastomas indicative of von Hippel-Lindau syndrome
Nurhuda Hendra Setyawan, Rachmat Andi Hartanto, Rusdy Ghazali Malueka, Ery Kus Dwianingsih, Dito Pondra Dharma
Radiology Case Reports.2024; 19(11): 5000. CrossRef - Phenotypic and Genotypic Features of a Chinese Cohort with Retinal Hemangioblastoma
Liqin Gao, Feng Zhang, J. Fielding Hejtmancik, Xiaodong Jiao, Liyun Jia, Xiaoyan Peng, Kai Ma, Qian Li
Genes.2024; 15(9): 1192. CrossRef - Case report: Hemangioblastoma in the brainstem of a dog
Kirsten Landsgaard, Samantha St. Jean, Stephanie Lovell, Jonathan Levine, Christine Gremillion, Brian Summers, Raquel R. Rech
Frontiers in Veterinary Science.2023;[Epub] CrossRef - Intramedullary hemangioblastoma of the thoracic cord with a microsurgical approach: A case report and literature review
Eduardo Cattapan Piovesan, Werner Petry Silva, Adroaldo Baseggio Mallmann, Felipe Severo Lanzini, Bruna Zanatta de Freitas, Francisco Costa Beber Lemanski, Charles André Carazzo
Surgical Neurology International.2023; 14: 137. CrossRef - Secondary Holocord Syringomyelia Associated With Spinal Hemangioblastoma in a 29-Year-Old Female
Eric Chun-Pu Chu, Edouard Sabourdy, Benjamin Cheong
Cureus.2023;[Epub] CrossRef - Belzutifan in adults with VHL-associated central nervous system hemangioblastoma: a single-center experience
Bryan J. Neth, Mason J. Webb, Jessica White, Joon H. Uhm, Pavel N. Pichurin, Ugur Sener
Journal of Neuro-Oncology.2023; 164(1): 239. CrossRef - Resection of Intramedullary Hemangioblastoma: Timing of Surgery and Its Impact on Neurological Outcome and Quality of Life
Michael Schwake, Sarah Ricchizzi, Sophia Krahwinkel, Emanuele Maragno, Stephanie Schipmann, Walter Stummer, Marco Gallus, Markus Holling
Medicina.2023; 59(9): 1611. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
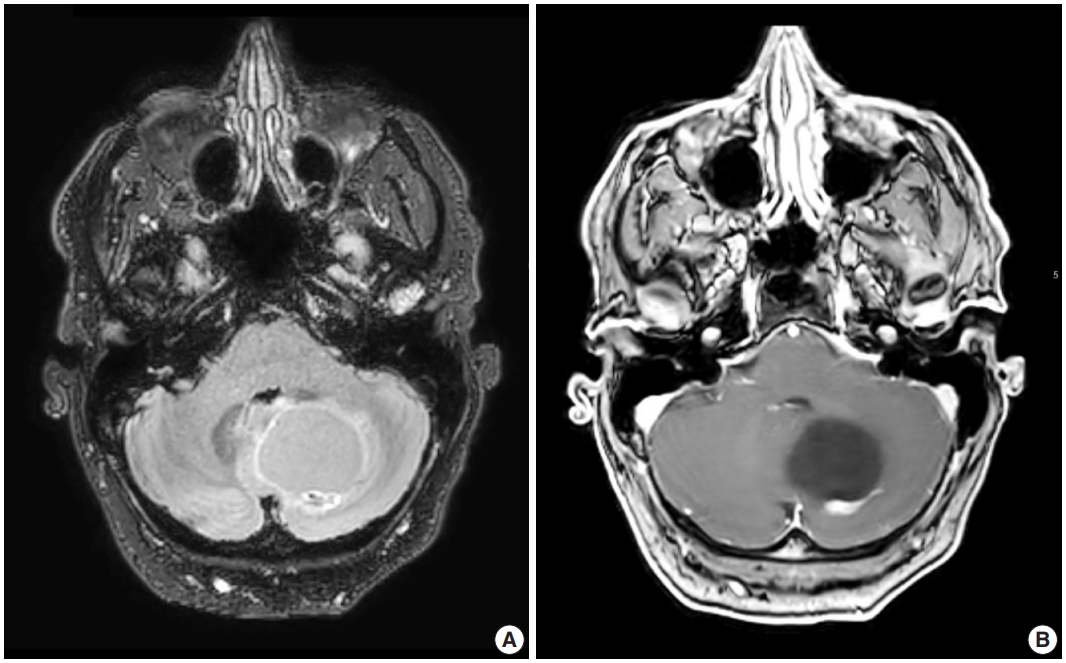
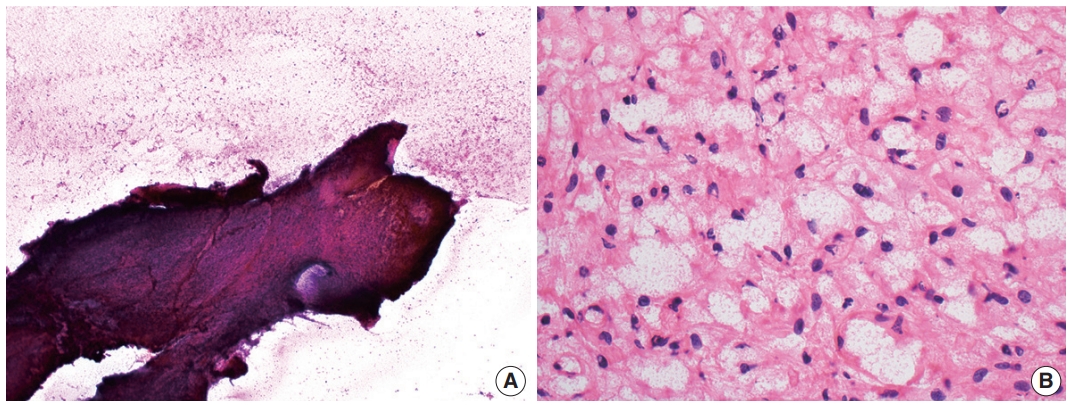
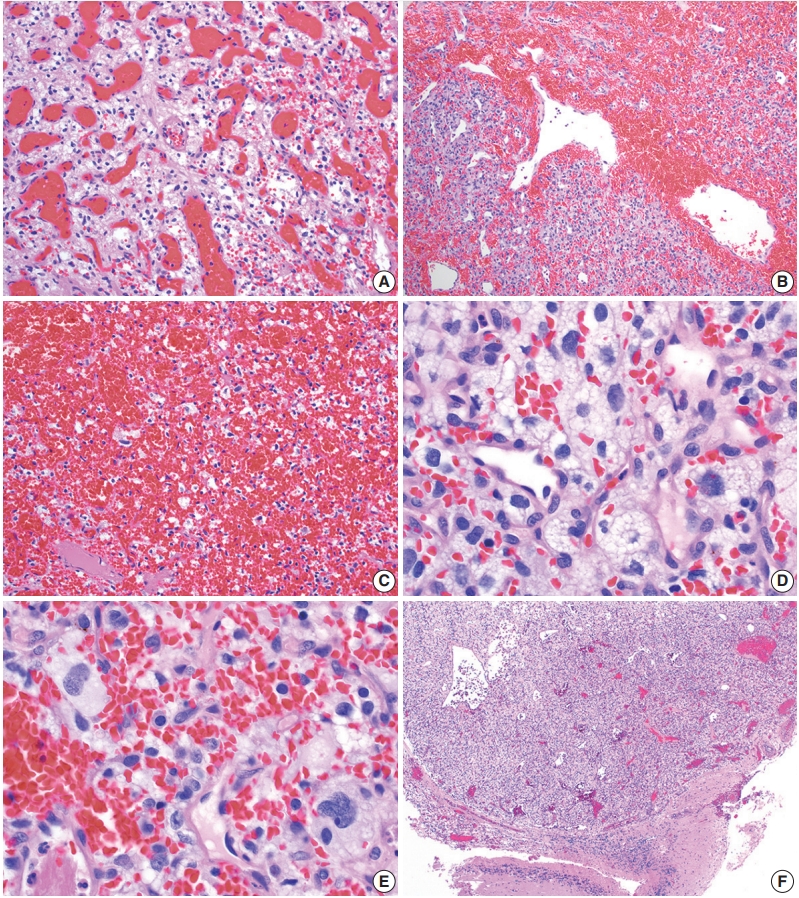
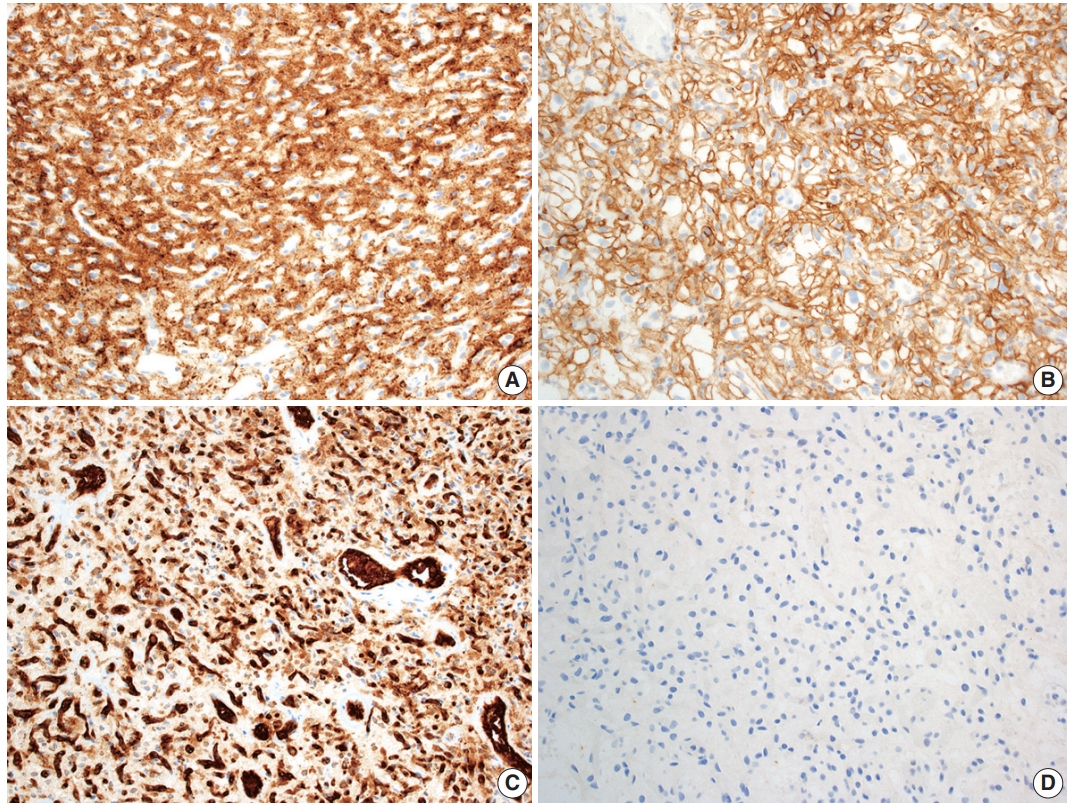




Fig. 1.
Fig. 2.
Fig. 3.
Fig. 4.
| Sporadic | Hereditary | |
|---|---|---|
| Proportion of total cases (%) | ~70 | ~30 |
| Mean age of onset (yr) | 47 | 29 |
| Sex (M:F) | 1–1.25:1 | 1–1.25:1 |
| No. of tumors | Single | Multiple |
| Localization | Cerebellum most common | Higher proportion of extracerebellar sites (e.g. spine) |
| Molecular alterations | Somatic loss of VHL detected in subset of cases | VHL loss of function with germline mutation and somatic alteration |
VHL, von Hippel-Lindau.

