 E-submission
E-submission
Search
- Page Path
- HOME > Search
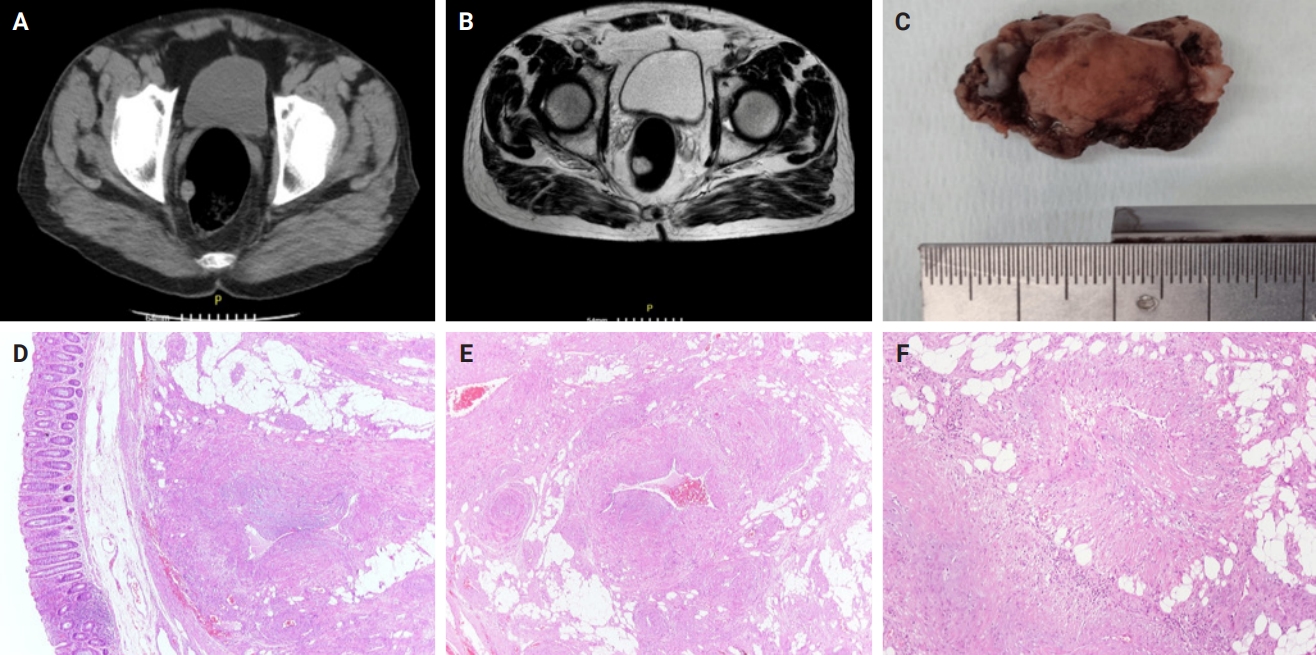
- Clinicopathological characteristics of digestive system angioleiomyomas: case report and literature review
- Georgios Kalliopitsas, Christos Topalidis, Constantine Halkias, Theodora Gkeka, Konstantinos Sapalidis, Triantafyllia Koletsa
- J Pathol Transl Med. 2025;59(6):453-459. Published online October 28, 2025
- DOI: https://doi.org/10.4132/jptm.2025.08.04
- 4,021 View
- 119 Download
-
 Abstract
Abstract
 PDF
PDF - Angioleiomyomas are benign soft tissue tumors originating from the vascular wall. Although angioleiomyomas mainly occur in extremities, followed by head, neck, and trunk, they can also be found throughout the digestive system and especially in the oral cavity. Herein, the fourth case of a rectal angioleiomyoma in the English literature is reported and the clinicopathological features of digestive system angioleiomyomas were investigated. In contrast to their soft tissue counterparts, digestive system angioleiomyomas mainly affect males at a slightly younger age. Angioleiomyomas are mainly asymptomatic and only rarely elicit pain. Clinicians consider angioleiomyomas infrequently and instead include more common soft tissue or epithelial tumors in their differential diagnosis. To prevent angiomyolipoma misdiagnosis, pathologists should exercise caution when examining an angioleiomyoma composed of adipose tissue, smooth muscle, and blood vessels. Pathologists, radiologists, and surgeons should be aware that angioleiomyomas can occur in the digestive system.
- Endobronchial Smooth Muscle Tumors: A Series of Five Cases Highlighting Pitfalls in Diagnosis
- Tripti Nakra, Aanchal Kakkar, Shipra Agarwal, Karan Madan, Suresh C Sharma, Deepali Jain
- J Pathol Transl Med. 2018;52(4):219-225. Published online July 11, 2018
- DOI: https://doi.org/10.4132/jptm.2018.05.16
- 11,791 View
- 115 Download
- 7 Web of Science
- 5 Crossref
-
Abstract
PDF
- Background
Primary endobronchial smooth muscle tumors (SMTs), which are extremely rare, include endobronchial leiomyomas and leiomyosarcomas. Clinically, SMTs present with signs and symptoms of bronchial obstruction, and lack specific radiological findings. Thus, histopathological examination is required for accurate diagnosis as well as for tumor grading. We examined the histomorphological and immunohistochemical features of endobronchial SMTs and highlighted pitfalls in diagnosis, particularly when using small biopsies.
Methods
Cases of primary endobronchial SMTs diagnosed at our Institute over the last 6 years (2012–2017) were retrieved from the departmental archives. Histopathological features and immunohistochemistry performed for establishing the diagnosis were reviewed.
Results
Five cases of SMTs occurring in endobronchial locations were identified. These included three cases of leiomyoma, and two cases of leiomyosarcoma. The age distribution of patients ranged from 13 to 65 years. Leiomyomas showed more consistent staining with smooth muscle markers (smooth muscle actin, desmin, and smooth muscle myosin heavy chain), while tumors of higher grade showed variable, focal staining, leading to erroneous diagnosis, especially on small biopsies.
Conclusions
The diagnosis of endobronchial SMTs relies on histopathological examination, for both confirmation of smooth muscle lineage and determination of the malignant potential of the lesion. Appropriate immunohistochemical panels including more than one marker of smooth muscle differentiation are extremely valuable for differential diagnosis from morphological mimics, which is necessary for instituting appropriate management. -
Citations
Citations to this article as recorded by
- Case report: Successful bronchoscopic interventional treatment of endobronchial leiomyomas
Yinfeng Wang, Yixiang Zhang, Ruirui Tong
Open Life Sciences.2024;[Epub] CrossRef - Pediatric endobronchial tumors with a mimicker: A case series
Kulwiwat Promsawasdi, Teerasak Phewplung
Pediatric Pulmonology.2024; 59(10): 2669. CrossRef - Smooth Muscle Conditions of the Chest
Matthew R. McCann, Lucas R. Massoth, Carlos A. Rojas, Yin P. Hung, John P. Lichtenberger, Gerald F. Abbott, Justin T. Stowell
Journal of Thoracic Imaging.2021; 36(5): 263. CrossRef - A Well-Defined Endobronchial Tumor in a 26-Year-Old Man
Christina Triantafyllidou, Petros Effraimidis, Mirjam Schimanke, Simone Ignatova, Anders Ringman, Susann Skoog, Farkas Vánky, Miklós Boros, Karin Cederquist
Chest.2021; 159(5): e313. CrossRef - Primary Pulmonary Leiomyoma
Mohammad Abu-Hishmeh, Gowthami Kobbari, Fouzia Shakil, Oleg Epelbaum
Journal of Bronchology & Interventional Pulmonology.2020; 27(4): e54. CrossRef
- Case report: Successful bronchoscopic interventional treatment of endobronchial leiomyomas
- A Rare Case of Angioleiomyoma Arising in the Subglottic Area to Upper Trachea of a Patient with Underlying Asthma
- Yeoun Eun Sung, Chin Kook Rhee, Kyo Young Lee
- J Pathol Transl Med. 2017;51(1):92-95. Published online August 22, 2016
- DOI: https://doi.org/10.4132/jptm.2016.06.21
- 10,613 View
- 114 Download
- 4 Web of Science
- 3 Crossref
-
Abstract
PDF
- Angioleiomyoma is a rare disease that is histologically characterized by smooth muscle cells arranged around vascular spaces. Although angioleiomyomas occur rarely in the head and neck region, they can cause various symptoms according the site involved. Here, we present a 44-yearold male patient with a 15-year history of asthma, who presented with recent onset of chest discomfort, globus sensation and throat pain. Medication was not effective in relieving his symptoms, and further evaluation revealed a polypoid ovoid mass, almost obstructing the airway at the border of the larynx and upper trachea on chest computed tomography. The mass was completely resected via a rigid bronchoscopy procedure. Histopathologic examination revealed that the excised mass was angioleiomyoma, which was immunohistochemically positive for smooth muscle actin and negative for desmin.
-
Citations
Citations to this article as recorded by- Angioleiomyoma of the Epiglottis Mimicking Epiglottic Hemangioma: Clinical Experience and Literature Review
Yang-Yang Bao, Xiao-Jie Shi, Li-Bo Dai, Yu Guo, Hong-Tian Yao, Shui-Hong Zhou
Ear, Nose & Throat Journal.2025; 104(3): NP125. CrossRef - Angioleiomyoma of the Larynx: A Case Report and Literature Review
Federica Perardi, Giuseppe Abbate, Leonardo R. Iannuzzelli, Rossella Contini, Manuela De Munari, Francesco G. Sciuto, Monica Leutner, Antonio Scotti
Ear, Nose & Throat Journal.2020; 99(10): 658. CrossRef - Flexible bronchoscopy and cryoextraction for critical airway obstruction caused by an endobronchial angioleiomyoma
Sumit Chatterji, Efrat Ofek, Tiberiu Shulimzon
Respirology Case Reports.2019;[Epub] CrossRef
- Angioleiomyoma of the Epiglottis Mimicking Epiglottic Hemangioma: Clinical Experience and Literature Review
- Extrapulmonary Lymphangioleiomyoma: Clinicopathological Analysis of 4 Cases
- Dae Hyun Song, In Ho Choi, Sang Yun Ha, Kang Min Han, Jae Jun Lee, Min Eui Hong, Yoon-La Choi, Kee-Taek Jang, Sang Yong Song, Chin A Yi, Joungho Han
- Korean J Pathol. 2014;48(3):188-192. Published online June 26, 2014
- DOI: https://doi.org/10.4132/KoreanJPathol.2014.48.3.188
- 10,720 View
- 65 Download
- 10 Crossref
-
Abstract
PDF
Background Lymphangioleiomyomatosis (LAM) is a slowly progressive neoplastic disease that predominantly affects females. Usually, LAM affects the lung; it can also affect extrapulmonary sites, such as the mediastinum, the retroperitoneum, or the lymph nodes, although these locations are rare. A localized form of LAM can manifest as extrapulmonary lesions; this form is referred to as extrapulmonary lymphangioleiomyoma (E-LAM). Due to the rare occurrence of E-LAM and its variable, atypical location, E-LAM is often difficult to diagnose. Herein, we report the clinicopathological information from four E-LAM cases, and also review previous articles investigating this disease.
Methods Four patients with E-LAM were identified at the Samsung Medical Center (Seoul, Korea) from 1995 to 2012. All E-LAM lesions underwent surgical excision.
Results All patients were females within the age range of 43 to 47 years. Two patients had para-aortic retroperitoneal masses, while the other two patients had pelvic lesions; two out of the four patients also had accompanying pulmonary LAM. In addition, no patient displayed any evidence of tuberous sclerosis. Histologically, two patients exhibited nuclear atypism with cytologic degeneration.
Conclusions E-LAM should be considered in the differential diagnosis of patients presenting with pelvic or para-aortic masses. We also conclude that further clinical and pathological evaluation is needed in patients with E-LAM and nuclear atypism.
-
Citations
Citations to this article as recorded by- Surgical Management of Solitary Extrapulmonary Lymphangioleiomyomatosis in the Mesentery: A Case Report
Jack Menzie, Chih C Kuan, Travis Ackermann, Yeng Kwang Tay
Cureus.2024;[Epub] CrossRef - Lymphangioleiomyomatosis with Tuberous Sclerosis Complex—A Case Study
Aleksandra Marciniak, Jolanta Nawrocka-Rutkowska, Agnieszka Brodowska, Andrzej Starczewski, Iwona Szydłowska
Journal of Personalized Medicine.2023; 13(11): 1598. CrossRef - A case of lymphangioleiomyomatosis with endometrial cancer diagnosed by retroperitoneoscopic para-aortic lymph node dissection
Aiko Ogasawara, Shogo Yamaguchi, Hiroaki Inui, Mieko Hanaoka, Daisuke Shintani, Sho Sato, Masanori Yasuda, Akira Yabuno
JAPANESE JOURNAL OF GYNECOLOGIC AND OBSTETRIC ENDOSCOPY.2022; 38(1): 158. CrossRef - Primary retroperitoneal PEComa: an incidental finding
Bárbara Monteiro Marinho, António Gâmboa Canha, Donzília Sousa Silva, José Davide Pinto Silva
BMJ Case Reports.2022; 15(11): e250466. CrossRef - Imaging Findings of Thoracic Lymphatic Abnormalities
Jingshuo (Derek) Sun, Thomas Shum, Fardad Behzadi, Mark M. Hammer
RadioGraphics.2022; 42(5): 1265. CrossRef - Extrapulmonary uterine lymphangioleiomyomatosis (LAM) and dysfunctional uterine bleeding: the first presentation of LAM in a tuberous sclerosis complex patient
Lucy Grant, Saliya Chipwete, San Soo Hoo, Anjali Bhatnagar
BMJ Case Reports.2019; 12(2): e226358. CrossRef - Summary of the Japanese Respiratory Society statement for the treatment of lung cancer with comorbid interstitial pneumonia
Takashi Ogura, Nagio Takigawa, Keisuke Tomii, Kazuma Kishi, Yoshikazu Inoue, Eiki Ichihara, Sakae Homma, Kazuhisa Takahashi, Hiroaki Akamatsu, Satoshi Ikeda, Naohiko Inase, Tae Iwasawa, Yuichiro Ohe, Hiromitsu Ohta, Hiroshi Onishi, Isamu Okamoto, Kazumasa
Respiratory Investigation.2019; 57(6): 512. CrossRef - Incidental lymphangioleiomyomatosis in the lymph nodes of gynecologic surgical specimens
Ikumi Kuno, Hiroshi Yoshida, Hanako Shimizu, Takashi Uehara, Masaya Uno, Mitsuya Ishikawa, Tomoyasu Kato
European Journal of Obstetrics & Gynecology and Reproductive Biology.2018; 231: 93. CrossRef - Solitary extrapulmonary lymphangioleiomyomatosis of the liver: A case report and literature review
Weiwei Fu, Yujun Li, Hong Li, Ping Yang, Xiaoming Xing
Experimental and Therapeutic Medicine.2016; 12(3): 1499. CrossRef - Incidental Pelvic and Para-aortic Lymph Node Lymphangioleiomyomatosis Detected During Surgical Staging of Pelvic Cancer in Women Without Symptomatic Pulmonary Lymphangioleiomyomatosis or Tuberous Sclerosis Complex
Joseph T. Rabban, Brandie Firetag, Ankur R. Sangoi, Miriam D. Post, Charles J. Zaloudek
American Journal of Surgical Pathology.2015; 39(8): 1015. CrossRef
- Surgical Management of Solitary Extrapulmonary Lymphangioleiomyomatosis in the Mesentery: A Case Report
- Primary Myxoid Leiomyoma of the Liver
- Hee Seung Choi, Chang Won Jung, Soo Youn Cho, Sang Bum Kim, Sunhoo Park
- Korean J Pathol. 2014;48(1):54-57. Published online February 25, 2014
- DOI: https://doi.org/10.4132/KoreanJPathol.2014.48.1.54
- 10,890 View
- 73 Download
- 2 Crossref
-
Abstract
PDF
Herein, we report a case of primary myxoid leiomyoma of the liver. A 60-year-old woman complained of upper abdominal fullness. Computed tomography showed a solid tumor (8 cm) in the liver. The patient underwent right hepatectomy and histological findings from the resected specimen revealed scattered bland spindle cells in a background of exuberant myxoid material. The tumor cells were immunoreactive for smooth muscle actin and desmin. No other lesions were found elsewhere in the body. Thus, the tumor was diagnosed as a primary myxoid leiomyoma of the liver.
-
Citations
Citations to this article as recorded by- Hepatic Myxoid Leiomyoma: A Very Rare Tumor
João Fraga, Rui Caetano Oliveira, Luigi Terracciano, Mário Rui Silva, Maria Augusta Cipriano
GE - Portuguese Journal of Gastroenterology.2020; 27(5): 352. CrossRef - A Firm Hepatic Mass Cannot Be Penetrated by US-Guided Needle Biopsy
Suk Hyun Jang, Sun Moon Kim, Jang Sihn Sohn, Ki Hyun Ryu, Hyung Bin Yuk
Clinical Ultrasound.2016; 1(2): 126. CrossRef
- Hepatic Myxoid Leiomyoma: A Very Rare Tumor
- Cotyledonoid Dissecting Leiomyoma of the Uterus with Intravascular Luminal Growth: A Case Study
- Na Rae Kim, Chan Yong Park, Hyun Yee Cho
- Korean J Pathol. 2013;47(5):477-480. Published online October 25, 2013
- DOI: https://doi.org/10.4132/KoreanJPathol.2013.47.5.477
- 14,626 View
- 81 Download
- 7 Crossref
-
Abstract
PDF
Here, we report the case of a 43-year-old female who was diagnosed with a cotyledonoid dissecting leiomyoma (CDL) of the uterus. CDL is a recently described and extremely rare variant of a benign leiomyoma that can grossly masquerade as a malignancy. The 13-cm sized tumor was located primarily on the extrauterine surface as an intrauterine continuity, which showed dark red, congested, bulbous protuberances. It was multinodular appearance, encasing the bilateral adnexae and the left iliac vein. Microscopically, the nodules were separated by extensive hydropic degeneration. The nodules were composed of cigar-shaped spindle cells with no mitosis, cellular pleomorphism or coagulation necrosis. They also showed an intravascular luminal growth pattern. CDL with intravascular growth was diagnosed after excluding intravascular leiomyomatosis, disseminated peritoneal leiomyomatosis, and benign metastasizing leiomyoma. The present case is the second reported case of CDL in Korea. Recognition of this rare and bizarre, malignancy-mimicking leiomyoma is crucial to prevent inappropriate treatment.
-
Citations
Citations to this article as recorded by- A Case of Cotyledonoid-Dissecting Leiomyoma - The Utility of Laparoscopic Biopsy and Gonadotropin-Releasing Hormone Analogs
Sayaka Kawashita, Akiko Nonoshita, Keisuke Iwasaki, Daisuke Nakayama
Clinical Pathology.2024;[Epub] CrossRef - Cotyledonoid dissecting leiomyoma with peritoneal dissemination
Hiroki Egashira, Hiroaki Ishida, Nobuyuki Hiruta, Akiko Takashima
BMJ Case Reports.2024; 17(9): e261937. CrossRef - Cotyledonoid dissecting leiomyoma of the uterus: a case report and review of the literature
Mahboobeh Chahkandi, Marzieh Ataei, Amir Reza Bina, Farnaz Mozayani, Ali Fanoodi
Journal of Medical Case Reports.2023;[Epub] CrossRef - Cotyledonoid Leiomyoma Clinical Characteristics, Imaging Features, and Review of the Literature
Francesca Buonomo, Sofia Bussolaro, Giorgio Giorda, Federico Romano, Stefania Biffi, Giuseppe Ricci
Journal of Ultrasound in Medicine.2021; 40(7): 1459. CrossRef - The Management of the Cotyledonoid Leiomyoma of the Uterus: A Narrative Review of the Literature
Francesca Buonomo, Sofia Bussolaro, Clarice de Almeida Fiorillo, Giorgio Giorda, Federico Romano, Stefania Biffi, Giuseppe Ricci
International Journal of Environmental Research and Public Health.2021; 18(16): 8521. CrossRef - Cotyledonoid dissecting leiomyoma of the uterus: A report of four cases and a review of the literature
TIANMIN XU, SHUYING WU, RULIN YANG, LIPING ZHAO, MINGXING SUI, MANHUA CUI, WEIQIN CHANG
Oncology Letters.2016; 11(4): 2865. CrossRef - COTYLEDONOID DISSECTING LEIOMYOMA (CDL) OF UTERUS MIMICKING MALIGNANCY: A CLINICAL DILEMMA
Roma Isaacs, Rupinder Kaur, Sunita Goyal
Journal of Evolution of Medical and Dental Sciences.2016; 5(57): 3973. CrossRef
- A Case of Cotyledonoid-Dissecting Leiomyoma - The Utility of Laparoscopic Biopsy and Gonadotropin-Releasing Hormone Analogs
- Gene Expression Profiles of Uterine Normal Myometrium and Leiomyoma and Their Estrogen Responsiveness In Vitro.
- Eun Ju Lee, Prati Bajracharya, Dong Mok Lee, Kyung Hyun Cho, Keuk Jun Kim, Young Kyung Bae, Mi Jin Kim, Ki Ho Lee, Hang Jin Kim, Gun Ho Song, Sang Sik Chun, Inho Choi
- Korean J Pathol. 2010;44(3):272-283.
- DOI: https://doi.org/10.4132/KoreanJPathol.2010.44.3.272
- 5,614 View
- 42 Download
- 3 Crossref
-
Abstract
PDF
- BACKGROUND
Uterine leiomyomas are common benign smooth muscle tumors among the reproductive aged-women. The research has been aimed to identify the differentially expressed genes between normal myometrium and leiomyoma and to investigate the effects of E2 on their expression.
METHODS
Gene microarray analysis was performed to identify the differentially expressed genes between normal myomerium and leiomyoma. The data was confirmed at protein level by tissue microarray.
RESULTS
Gene microarray analysis revealed 792 upregulated genes in leiomyoma. Four genes (tropomyosin 4 [TPM4], collagen, type IV, alpha 2 [COL4alpha2], insulin-like growth factor binding protein 5 [IGFBP5], tripartite motif-containing 28 [TRIM28]) showed the most dramatic upregulation in all leiomyoma samples. Tissue microarray analyses of 262 sample pairs showed significantly elevated expression of TPM4, IGFBP5, estrogen receptor-alpha, and progesterone receptor (PR) protein in leiomyoma from the patients in their forties, COL4alpha2 in the forties and fifties age-groups, and TRIM28 in the thirties age-group. PR, insulin-like growth factor 1 (IGF-1), IGF-1 receptor (IGF-1R) and IGFBP5 were induced by E2 in in vitro culture of tissue explants from which cells migrated throughout the plate. Among these, PR, IGF-1, IGFBP5 genes showed higher expression in tissue compared to cells-derived from tissue in leiomyoma and IGF-1R in leiomyoma cell.
CONCLUSIONS
This observation implies the importance of the whole tissue context including the cells-derived from tissue in the research for the understanding of molecular mechanism of leiomyoma. Here, we report higher expression of TRIM28 in leiomyoma for the first time and identify E2-responsive genes that may have important roles in leiomyoma development. -
Citations
Citations to this article as recorded by- In vivo mechanisms of uterine myoma volume reduction with ulipristal acetate treatment
Guillaume E. Courtoy, Jacques Donnez, Etienne Marbaix, Marie-Madeleine Dolmans
Fertility and Sterility.2015; 104(2): 426. CrossRef - Common fibroid-associated genes are differentially expressed in phenotypically dissimilar cell populations isolated from within human fibroids and myometrium
Sarah J Holdsworth-Carson, Marina Zaitseva, Jane E Girling, Beverley J Vollenhoven, Peter A W Rogers
Reproduction.2014; 147(5): 683. CrossRef - Complex networks of multiple factors in the pathogenesis of uterine leiomyoma
Md Soriful Islam, Olga Protic, Piergiorgio Stortoni, Gianluca Grechi, Pasquale Lamanna, Felice Petraglia, Mario Castellucci, Pasquapina Ciarmela
Fertility and Sterility.2013; 100(1): 178. CrossRef
- In vivo mechanisms of uterine myoma volume reduction with ulipristal acetate treatment
- Angioleiomyoma of the Nasal Cavity: A Case Report.
- Su Jin Kim, Sook Hee Hong, Mee Sook Roh
- Korean J Pathol. 2004;38(3):181-183.
- 2,368 View
- 15 Download
-
Abstract
PDF
- Angioleiomyoma of the sinonasal area is an extremely rare benign neoplasm. To the best of our knowledge, only 26 cases have been described. Here, we report a case of angioleiomyoma arising in the nasal cavity of a 60-year-old woman. Microscopically, the tumor consisted of proliferating smooth muscle cells punctuated with thick-walled vessels with slit-like lumina. The tumor was negative for estrogen and progesterone receptor by immunohistochemical study. Further studies are needed to clarify whether the growth of this tumor is sex steroid-dependent.
- Atypical (Bizarre) Leiomyoma of the Prostate: A Case Report.
- Sung Rim Kim, Sang Yong Song, Geunghwan Ahn, Han Yong Choi
- Korean J Pathol. 2001;35(2):172-175.
- 2,512 View
- 21 Download
-
Abstract
PDF
- Atypical (bizarre) leiomyoma of the prostate is a very rare neoplasm. Five cases have been reported in English medical literature. A 60-year-old Korean man with a history of prostatism and slightly elevated serum prostate specific antigen was presented. Microscopically, the transurethral resection specimen consisted of a proliferation of hypercellular spindle cells with intersecting bundles. The nuclei of the tumor cells showed marked pleomorphism and hyperchromasia with occasional multinucleated giant cells. Mitoses were seen in areas of up to 2 per 10 high power fields, but there was no evidence of atypical ones. The tumor cells were immunoreactive against anti-smooth muscle actin and desmin antibodies. The proliferative index (10.0%) of the atypical leiomyoma lay between that of a benign smooth muscle and that of a leiomyosarcoma of the prostate. Flow cytometry showed a diploid pattern with an elevated S phase fraction. To the best of our knowledge, this is the first demonstration of atypical leiomyoma of the prostate in a Korean man.
- Leiomyoma of the Female Urethra: A case report.
- Kyoung Mee Kim, Anhi Lee, Sang In Shim
- Korean J Pathol. 1995;29(5):684-686.
- 2,316 View
- 10 Download
-
Abstract
- Leiomyomas are benign tumors of smooth muscle origin and are very rare in the female genital tract. To date, approximately 35 cases of urethral leiomyoma have been reported in the literature. A 34-year-old woman presented with a 3-year history of a mass at the urethral meatus. Physical examination showed 2 x 1.5 cm lump at the urethral meatus, posterior lip. Histologically the tumor was mainly composed of benign cigar shaped smooth muscle izells which were arranged in interlacing fascicles without cellular atypia or mitosis. Immunohistochemistry confirmed leiomyoma with positive staining for vimentin, desmin and muscle specific actin.
- Leiomyoma of the Ovary A report of two cases.
- Jeong Hae Kie, Tai Seung Kim, Dong Hwan Shin
- Korean J Pathol. 1999;33(7):529-532.
- 2,105 View
- 15 Download
-
Abstract
PDF
- Ovarian leiomyoma is a rare form of the ovarian mesenchymal neoplasm and about 50 cases have been reported in the literature. It is believed that many cases may go unnoticed because they are usually small in size and frequently mistaken for the more common fibroma or fibrothecoma. Its origin is still controversial and many possibilities are considered including the smooth muscle in the blood vessel wall of the hilum or the multipotential ovarian stromal cell. Herein we describe two cases of ovarian leiomyoma with its characteristic histologic finding.
- Diffuse Leiomyomatosis of the Uterus: A Brief Case Report.
- Su Jin Kim, Mee Sook Roh
- Korean J Pathol. 2005;39(1):63-65.
- 3,742 View
- 97 Download
-
Abstract
PDF
- Diffuse leiomyomatosis of the uterus is a rare condition that is distinguished from the uterine leiomyoma due to the diffuse involvement of the myometrium by numerous, ill-defined, smooth muscle nodules. We present here a case of diffuse uterine leiomyomatosis in a 34-year-old woman. The hysterectomy revealed a symmetrically enlarged uterus containing numerous, small, ill-defined leiomyomatous nodules. Microscopically, the nodules were composed of compact fascicles and interweaving bundles of uniform benign smooth muscle cells. On the immunohistochemical staining, the progesterone receptor level was higher in the leiomyomatosis than in the adjacent normal myometrial tissue, but the estrogen receptor level and Ki-67 labeling index were equal in both areas. At the twelve months follow-up, this patient has been doing very well with no evidence of pelvic or intraabdominal recurrence of disease.
- Lipoleiomyoma of the Uterus: A case report.
- Myung Sook Kang, Young Hee Maeng, Jae Hoon Park, Yun Wha Kim, Ju Hee Lee, Moon Ho Yang
- Korean J Pathol. 1993;27(5):535-537.
- 2,210 View
- 27 Download
-
Abstract
PDF
- A rare case of uterine lipoleiomyoma is reported with presentation of computed tomography, histomorphologic and immunohistochemical findings. This tumor is predominantly lipomatous with an admixture of smooth muscle fiber and hyalinized fibrous tissue. Immunohistochemical study revealed a positive reaction of S-100 protein in fat cells and desmin in smooth muscle fibers. Its histogenesis also has been discussed.
- Immunohistochemical Analysis of Estrogen Receptors and Progesterone Receptors in Leiomyoma of Uterus Compared with PCNA Index.
- Jung Ran Kim
- Korean J Pathol. 1996;30(2):140-149.
- 2,735 View
- 49 Download
-
Abstract
PDF
- Estrogen receptor(ER) and progesterone receptor(PR) were studied immunohistochemically using specific antireceptor monoclonal antibodies in leiomyomas and myometrium from same patients from 38 women in various stages of the menstrual cycle, menopause and pregnancy. Two postpartum uteri are also included. Immunohistochemical localization was quantified as to intensity of staining and tissue distribution, and the results were compared with those of PCNA index. In all samples, ER and PR localized within the nuclei of target cells. The histochemical score of ER in leiomyoma was significantly greater than that found in myometrium. But ER in leiomyoma was expressed in cyclic fashion(r=0.45, P=0.006), like as in myometrium, throughout the menstrual cycle, paralleled by a concomitant, though delayed. In contrast, PR content constantly maintained in myometrium and leiomyoma throughout menstrual cycle, and there was no significant difference between them. However, leiomyoma and myometrium of pregnancy showed a significant reduction in the amount of ER and PR localized. PCNA index in leiomyoma(14.9+/-24.4) was also significantly higher than that found in myometrium(2.1+/-3.3). The index declined throughout the secretory phase. The leiomyoma had increased PCNA index during pregnancy, while the increasing rate in leiomyoma was lower than that of myometrium. The growth potential of leiomyomas is appearently higher than that of myometrium under the high progesterone level. The most of neoplasm with high PCNA index(10 above) contained absolute or relative abundant PR or ER content. Alteration of receptor content may be an important mechanism in steroid dependent growth of leiomyoma and may provide information useful in the clinical management of this neoplastic disorder.
- CD34 Antigen Expression in Gastrointestinal Stromal Tumors.
- Sun Hee Sung, Min Sun Cho, Woon Sup Han
- Korean J Pathol. 1997;31(11):1166-1171.
- 4,690 View
- 13 Download
-
Abstract
- Gastrointestinal stromal tumor (GIST) is known as considerable controversal tumor about it's histogenesis, differentiation and biologic behavior. It is traditionally regarded as smooth muscle tumor. To evaluate and clarify the origin of tumor, we performed immunohistochemical study of 23 cases of GIST on CD34 antigen, alpha-smooth muscle actin, S-100 protein, and compared the result with 4 cases of typical leiomyoma of GI tract. The results were as follows. CD34 antigen expression was noted in 21 cases (91.3%) of GIST, while typical leiomyoma was all negative. There were no difference of CD34 expression according to the biologic behavior. However, it's staining pattern was significantly different (p<0.05). Focal or multifocal expression was dominant in benign GIST (58.3%), while diffuse expression was dominant in malignant GIST (80%). Actin was expressed in 5 cases of benign GIST (38.5%) and 1 of malignant GIST (16.7%) focally. All typical leiomyoma showed diffuse strong positivity on alpha-smooth muscle actin. S-100 protein was expressed in 2 cases of benign GIST (16.7%) only. The pattern of CD34 expression was focal in the actin or S-100 protein positive cases. In conclusion CD34 antigen is useful marker in the separation of GIST, from typical smooth muscle tumor. Also it suggest that most GISTs are histogenetically primitive mesenchymal cell origin. However, CD34 expression was unrelated with biologic behavior of GIST.
- Leiomyoma of the Ovary: A Case Report.
- Hye Kun Oh, Yeun Kyung Lee, Sung Chul Lim
- Korean J Pathol. 2002;36(1):59-61.
- 2,385 View
- 29 Download
-
Abstract
PDF
- We present a case of ovarian leiomyoma without related clinical symptoms in a 68-year-old woman. Leiomyoma arising primarily in the ovary is rare. However, it is believed that there are actually more cases than those reported because this condition is usually mistaken for a fibrothecoma or parasitic leiomyoma. Most cases previously reported were incidentally presented and coexisted with other ovarian lesions. The present case was characterized by a 9 cm, round lobulated mass that totally replaced the left ovary without uterine leiomyoma or coexisting ovarian lesions.
- Leiomyoma of the Lung: A case report.
- Seung Yeon Ha, Yung Suk Lee, Won Bo Cho, In Sun Kim
- Korean J Pathol. 1993;27(6):673-675.
- 2,205 View
- 20 Download
-
Abstract
PDF
- We present a 37-year-old male who was found to had mas within the bronchus. This patients was admitted for the evaluation of cough. Chest CT scan showed endobronchial mass in the bifurcation of LUL and LLL bronchus. The left lower lobe was atelectatic. Lobectomy of the left lower lobe was done. On opening of the bronchus, there was a 2x1x1 cm sized endobronchial mass. Histologically, the mass was smooth muscle tumor composed of densely packed eosinophilic spindle cells in interlacing bundles with pale elongated nuclei covered by bronchial epithelium. On immunohistochemical staining, the tumor cells were positive for desmin. Ultrastructurally, the tumor cells exhibited numberous cytoplasmic microfilaments with focal densities, pinocytotic vesicles, and a thick basal lamina.
- Immunohistochemical and Ultrastructural Studies of Gastric Smooth Muscle Tumor.
- Hyang Mi Ko, Kyung Soo Kim, Jae Hyuk Lee, Woo Sik Juhng, Sang Woo Juhng
- Korean J Pathol. 1996;30(3):245-254.
- 2,381 View
- 19 Download
-
Abstract
PDF
- To evaluate the differentiation status of smooth muscle in gastric stromal tumors which were negative for S-100 protein, immunohistochemistry using desmin, actin, myosin and vimentin was performed in 14 cases of gastric smooth muscle tumors. Ultrastructural Examination was also performed. For comparison a case of leiomyoma of the esophagus, a case of the sigmoid colon, 10 cases of the uterus were also examined. The results obtained were as follows. All gastric smooth muscle tumors showed vimentin-positivity. Six of 14 gastric smooth muscle tumors, (5 of 8 leiomyoma and 1 of 4 leiomyosarcoma) showed positivity for desmin, actin, and myosin(42.9%). All esophageal, colonic, and uterine leiomyomas showed diffuse positive reaction for desmin, actin, and myosin. Vimentin positivity was also noted in leiomyoma of the colon and uterus. Ultrastructurally, a few cells in the gastric stromal tumors had scattered microfilaments with dense bodies, subplasmalemmal dense plaques, and micropinocytic vesicles. However, most of the tumor cells did not have any of the ultrastructural features of smooth muscle differentiation. Leiomyomas of the esophagus and uterus showed many cytoplasmic microfilaments with dense bodies. These results suggest that most of the benign and malignant tumor cells of gastric stromal tumors have features of the undifferentiated cells, immunohistochemically as well as ultrastructurally, although a few cells have. It is speculated that most gastric stromal tumors may have lost their smooth muscle differentiation.
- Leiomyoma of the Urinary Bladder.
- Kye Weon Kwon, Hee Jung Ahn, Yoon Jung Choi, Young Kwon Hong, Jae Seop Shin
- Korean J Pathol. 1997;31(12):1320-1323.
- 2,342 View
- 19 Download
-
Abstract
PDF
- Leiomyoma is commonly found in the female genital tract, but occurrence in the urinary bladder is very rare with only 235 cases reported in the literature. These tumors have been classified as intravesical (63%), intramural (7%) and extravesical (30%) depending on the direction of the growth. We report a case of intravesical leiomyoma of the urinary bladder in a 36 year-old woman who exhibited dysuria and urinary retention. The gross and microscopical findings of leiomyoma of the bladder are similar to those of the uterus. Immunohistochemical stains for estrogen receptor (ER) and progesterone receptor (PR) revealed diffuse nuclear staining in smooth muscle cells, supporting the hypothesis of hormonal influence in tumorigenesis.
- Uterine Leiomyomas with Perinodular Hydropic Degeneration: A Report of Two Cases.
- Sung Nam Kim, Jaejung Jang, Kyu Rae Kim
- Korean J Pathol. 2002;36(4):257-261.
- 2,888 View
- 47 Download
-
Abstract
PDF
- Hydropic degeneration is a frequent degenerative change in otherwise typical uterine leiomyomas. Very rarely, however, a significant amount of edema fluid accumulates around the fascicles of neoplastic smooth muscle bundles and forms the characteristic multinodular growth pattern that is called perinodular hydropic degeneration of leiomyoma (PHDL). The gross findings, showing a vague worm-like appearance and very rarely having an extrauterine extension, and the microscopic features, showing perinodular retraction artifacts forming pseudovascular spaces, make it difficult to differentiate the tumor from intravenous leiomyomatosis or myxoid leiomyosarcoma. We described two cases of leiomyomas showing perinodular hydropic degeneration (PHD), a condition that has rarely been described in English literature, and discussed the mechanism of forming "extrauterine extension" or cotyledonoid features. One of our cases showed the typical features of cotyledonoid dissecting leiomyoma, the other showed those of intramural dissecting leiomyoma. An awareness of the gross and microscopic findings of PHDL is important not to overdiagnose a benign smooth muscle neoplasm as a more aggressive type of tumor. It is thought that intramural dissecting leiomyoma, cotyledonoid dissecting leiomyoma, and PHDL are not distinct, but closely related subtypes showing different phases of evolutionary changes.
- Pulmonary Lymphangioleiomyomatosis: A case report.
- Won Bo Jo, Nam Hee Won, Seung Yong Paik, Hae Kyung Ahn
- Korean J Pathol. 1991;25(3):269-274.
- 2,595 View
- 24 Download
-
Abstract
PDF
- Lymphangioleiomyomatosis(LAM) is a rare disease of women of child-bearing age in which there is progressive hyperplasia of atypical smooth mucle along lymphatics in the lung, and/or axial lymphatics in the thorax and abdomen, resulting in honeycombing of lung. Interestingly there has been a speculation that it represents a forme furste or incomplete expression of tuberous sclerosis complex. This is based on the observation that patients with tuberous sclerosis can manifest pulmonary lesions indistinguishable from LAM. We report a case of LAM occuring in a 39-year-old female, who complained of recurrent pneumothorax, chest pain and shortness of breath. Three years ago, the patient had right nephrectomy under the diagnosis of ruptured angiomyolipoma. A X-ray film of the chest showed honeycombing with a diffusely reticulonodular pattern and cyst-like spaces. She had a characteristic facial appearance of adenoma sebaceum, which her father and uncle had. Microscopically, the lung showed a marked smooth muscle proliferation around the slit-like lymphatic spaces and also some respiratory bronchioles.
- Leiomyoma of the Skin: clinicopathological study of 19 cases.
- Seok Jin Kang, Sun Moo Kim
- Korean J Pathol. 1996;30(6):515-522.
- 2,300 View
- 19 Download
-
Abstract
PDF
- Nineteen cases of leiomyoma of the skin were examined clinicopathologically. This group included 12 cases of angioleiomyoma, 5 cases of solitary piloleiomyoma, and 2 cases of multiple piloleiomyomas. 1) All twelve angioleiomyomas occured as solitary lesion in the extremities. There was a preponderance in females with a ratio of 2:1. The ages of patients ranged from 24 to 80 years and only one was below the age of 30 years. Six tumors were either painful or tender. Nine tumors in subcutaneous fat were shelled out at surgery. All tumors did not exceed 4cm in diameter. Histologically they could be separated into ten cases of the solid type and two cases of venous type according to Morimoto's classification. Although actin or desmin was easily detected in all tumors, the diagnosis was better made using a combination of hematoxylin-eosin and Masson trichrome stains. 2) Five cases of solitary piloleiomyoma were slow-growing intradermal nodules. The ages of patients ranged from 10 to 77 years. All five cases were female. The lesions were located on the extremities, back and sholuder. Pain or tenderness was present in 3 cases among these tumors. Histologically, all tumors were characterized by subtle poorly circumscribed proliferation of benign smooth muscle in the dermis. 3) Two multiple piloleiomyomas from two female patients, aged 50 and 40 years, were situated on the shoulder and thigh, respectively. Pain was induced by change of temperature in the shoulder lesion. Histologically they were identical to the solitary piloleiomyoma.
- Pulmonary Lymphangioleiomyomatosis: Pathologic Analysis of Eight Korean Cases.
- Seung Sook Lee, Jeong Wook Seo, Eul Keun Ham, Yong Il Kim, Nam Hee Won, Jung Gi Im, Young Soo Shim
- Korean J Pathol. 1994;28(4):358-367.
- 2,235 View
- 15 Download
-
Abstract
PDF
- Histopathology of pulmonary lymphangioleiomyomatosis(LAM) is studied using four new cases and six previously reported cases, which include two cases without definite evidence of LAM. The important diagnostic features of this lesion were nodular proliferation of immature smooth muscle and cleft or cyst formation within the nodules of smooth muscle cells. The nuclei of the smooth muscle cells were bigger than those of blood vessels or fibrotic lung, and the direction of nuclei was irregular. The lung parenchyma showed little inflammatory change but there were multiple air cysts with smooth muscle nodules at their margin. There were two cases with exuberant proliferation of smooth muscle nodules and two cases with papilliferous projections of the cells into lymphatic lumen. Whereas, three cases had only a few small slender nodules of smooth muscle cells at the margin of air cyst. The lymphatic lumen with smooth muscle nodules is dilated in four cases but other four cases show collapsed lumen. Pulmonary hemorrhage and hemosiderosis were prominent in three cases. There were variety of histology in terms of the cellularity of smooth muscle nodules, the size of the lymphatic lumen and the degree of pulmonary destruction, which may have significance on the clinical presentation and prognostication.
- Uterine Leiomyoma with Massive Lymphocytic Infiltration.
- Won Mi Lee, Moon Hyang Park
- Korean J Pathol. 2003;37(1):71-73.
- 2,692 View
- 44 Download
-
Abstract
PDF
- Uterine leiomyoma with massive lymphocytic infiltration is known to be associated with Gona-dotropin releasing hormone (GnRH) agonist treatment. The lymphocytic cells in those cases were composed predominantly of T-lymphocytes. We report an unusual case of uterine leiomyoma with massive lymphocytic infiltration, composed predominantly of B-lymphocytes, without a history of GnRH agonist treatment. A 59-year-old woman underwent a transvaginal hysterectomy for uterine leiomyomas. Microscopically, the leiomyoma showed a massive infiltration of the lymphocytes, histiocytes, and also showed scattered plasma cells and many lymphoid follicles. The lymphocytic infiltrates were confined to the leiomyoma. These lymphocytic cells mainly represented the B-cell phenotype. She had no history of GnRH agonist treatment. To the best of our knowledge, This is the first reported case in Korea.
- A Clinicopathologic Study of 53 Gastrointestinal Mesenchymal Tumors.
- Young Kyung Bae, Dong Sug Kim, Mi Jin Gu, Joon Hyuk Choi, Mi Jin Kim, Young Jin Kim, Won Hee Choi, Sun Kyo Song, Koing Bo Kwun
- Korean J Pathol. 2000;34(11):909-918.
- 2,428 View
- 25 Download
-
Abstract
PDF
- The gastrointestinal mesenchymal tumors (GIMTs) form a heterogenous group with controversy centering on both the cell of origin and the prediction of clinical behavior. They include a small group of tumors with mature smooth muscle or Schwann cell differentiation and a larger group with inconsistent or no evidence of differentiation. Tumors in the latter are now referred to as gastrointestinal stromal tumors (GISTs). A clinicopathologic and immunohistochemical study was performed on 53 cases of GIMTs to identify cellular differentiation and predictors of clinical behavior. Fifty three cases of GIMTs could be histologically and immunophenotypically divided into three categories, 6 leiomyomas (11.3%), 4 schwannomas (7.6%), and 43 GISTs (81.1%). All leiomyomas (SMA desmin ) and schwannomas (S-100 ) were located in stomach and negative for CD34 and CD117. Thirty nine cases of GISTs were either CD34 (n=26) or CD117 (n=23) immunoreactive. Of these 39 GISTs, 26 were negative for myoid (SMA, desmin) and neural marker (S-100), 10 SMA desmin-S-100-, two SMA-desmin-S-100 , and one SMA desmin-S-100 . Two out of 4 GISTs, which were negative for CD34 and CD117, were immunohistochemically considered leiomyosarcoma (SMA desmin ). GISTs of small intestine had a tendency to be malignant than those of stomach. Pathologic grade of GISTs was not correlated with cellular differentiation. In 29 GISTs with clinical follow-up information, tumor size, mitotic counts, Ki-67 labelling index, tumor necrosis, mucosal invasion, and CD34 expression were significantly correlated with metastasis/recurrence.
- Diffuse Leiomyomatosis of the Esophagus: A case report.
- Ok Jun Lee, Hwa Sook Jeong, Jong Myeon Hong, Ro Hyun Sung
- Korean J Pathol. 1996;30(12):1159-1162.
- 1,946 View
- 14 Download
-
Abstract
PDF
- Diffuse leiomyomatosis of the esophagus is a rare condition and usually extends from the mid-esophagus to the proximal third of the stomach. Macroscopically, there is a marked diffuse thickening of the esophageal wall, with or without nodularity, predominantly affecting the circular muscle coat. Microscopically, the disorder is characterized by the loss of the normal orientation of the smooth muscle fibers of all three layers. We report a case in a 37-year-old woman which was incidentally discovered at exploratory thoracotomy.
- Yellowish Degeneration of Uterine Leiomyomas: Light Microscopic and Ultrastructural Observations.
- So Dug Lim, Joo Ryung Huh, Yong Il Kim
- Korean J Pathol. 1995;29(2):221-227.
- 4,562 View
- 140 Download
-
Abstract
PDF
- We reviewed five cases of Uterine leiomyomas, each with a conspicuously, yellow cut surface, among 198 consecutive cases of surgically removed uterine leiomyomas. Their gross findings were not significantly different from ordinary leiomyomas except for their pale to bright Yellowish cut surface. Microscopically, multiple small clusters of clear cells were widely scattered in otherwise hypercellular leiomyornas in 4 of the 5 cases. Of those, one case gave a positive reaction of Oil-Red O stain. Ultrastructurally, clear cells corresponded to the degenerating smooth muscle cells with intracytoplasmic lipid vacuoles. The rest of cells showed myofibers undergoing varying degrees of degeneration. Focal accumulation of foamy histiocytes was associated with carneous degeneration in one case. We conclude that the yellowish leiomyoma of the uterus seems, in part, to reflect accumulation of a lipid substance in degenerating hypercellular leiornyoma, or possibly collections of xanthoma cells in secondary degeneration.
 First
First Prev
Prev


