 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 52(5); 2018 > Article
-
Case Study
Ovarian Gynandroblastoma with a Juvenile Granulosa Cell Tumor Component in a Postmenopausal Woman: A Case Report and Literature Review -
Nu Ri Jang
 , Dae Hyung Lee1, Eun Jung Jang2, Young Kyung Bae, Jina Baek, Min Hye Jang
, Dae Hyung Lee1, Eun Jung Jang2, Young Kyung Bae, Jina Baek, Min Hye Jang -
Journal of Pathology and Translational Medicine 2018;52(5):344-348.
DOI: https://doi.org/10.4132/jptm.2018.06.28
Published online: July 17, 2018
Department of Pathology, Yeungnam University School of Medicine, Daegu, Korea
1Department of Gynecology and Obstetrics, Yeungnam University School of Medicine, Daegu, Korea
2Department of Pathology, Fatima Hospital, Daegu, Korea
- Corresponding Author Min Hye Jang, MD, PhD Department of Pathology, Yeungnam University School of Medicine, 170 Hyeonchung-ro, Nam-gu, Daegu 42415, Korea Tel: +82-53-640-6753 Fax: +82-53-622-8432 E-mail: clodious@naver.com
• Received: May 15, 2018 • Revised: June 25, 2018 • Accepted: June 28, 2018
© 2018 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Gynandroblastoma is an extremely rare sex cord-stromal tumor with both female (granulosa cell tumor) and male (Sertoli-Leydig cell tumor) elements. Juvenile granulosa cell tumors are also very rare and are so named because they usually occur in children and adolescents. A 71-year-old woman with right upper quadrant abdominal pain visited our hospital. Pelvic computed tomography showed a large multilocular cystic mass, suspected to be of ovarian origin. We performed a total abdominal hysterectomy (total abdominal hysterectomy was performed) with bilateral salpingo-oophorectomy. A 13-cm multilocular cystic mass with serous fluid was observed in her right ovary. Upon microscopic examination, the solid component of the mass showed both Sertoli-Leydig cell and juvenile granulosa cell differentiation, which we diagnosed as gynandroblastoma. Gynandroblastoma with a juvenile granulosa cell tumor component is extremely rare and, until now, only six cases have been reported in the English literature. We report the first gynandroblastoma with a juvenile granulosa cell tumor component diagnosed in an elderly patient, along with a literature review.
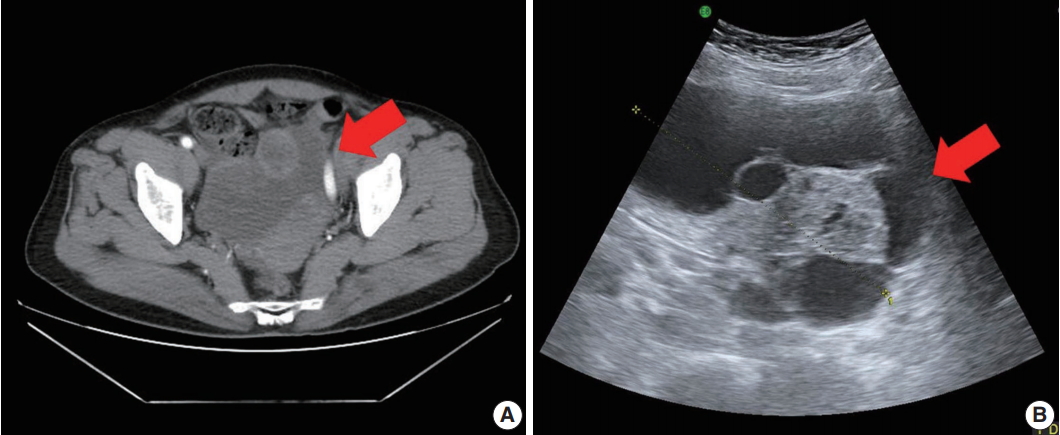
- A 71-year-old woman (gravida 6, para 4) with acute abdominal pain in the right lower quadrant visited our hospital’s emergency room. Computed tomography revealed a large multiloculated cystic mass in the pelvic cavity (Fig. 1A). Subsequent ultrasonography revealed a cystic mass of approximately 13 cm in the right ovary (Fig. 1B). The patient had no family history of genetic syndromes and no other underlying diseases. She experienced menarche at age 16 and menopause at age 53. She had no history of symptoms associated with endocrine manifestations, i.e., neither estrogenic nor androgenic changes, such as endometrial hyperplasia, abnormal endometrial bleeding, oligomenorrhea, infertility, or virilization.
- Her preoperative serum carbohydrate antigen (CA) 19-9 (19.48 U/mL) and CA125 (7.45 U/mL) levels were within normal ranges. Taking into consideration the preoperative diagnosis of torsion of the right ovarian tumor, the patient underwent total abdominal hysterectomy and bilateral salpingo-oophorectomy.
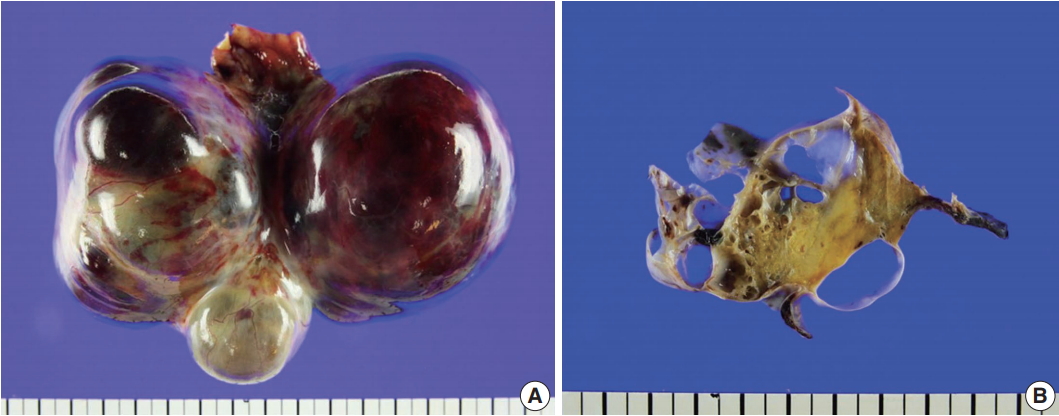
- On gross examination of the right ovary, a 13.0 × 7.5 × 6.5-cm multilocular ovarian cyst with a smooth and glistening surface was observed (Fig. 2A). The cyst was filled with serosanguinous fluid, and the inner surface had a mainly smooth appearance. In the cystic lumen, a 3.7 × 1.8-cm solid yellowish mass was observed (Fig. 2B). There were no remarkable findings upon gross examination of the uterus, fallopian tubes, and left ovary.
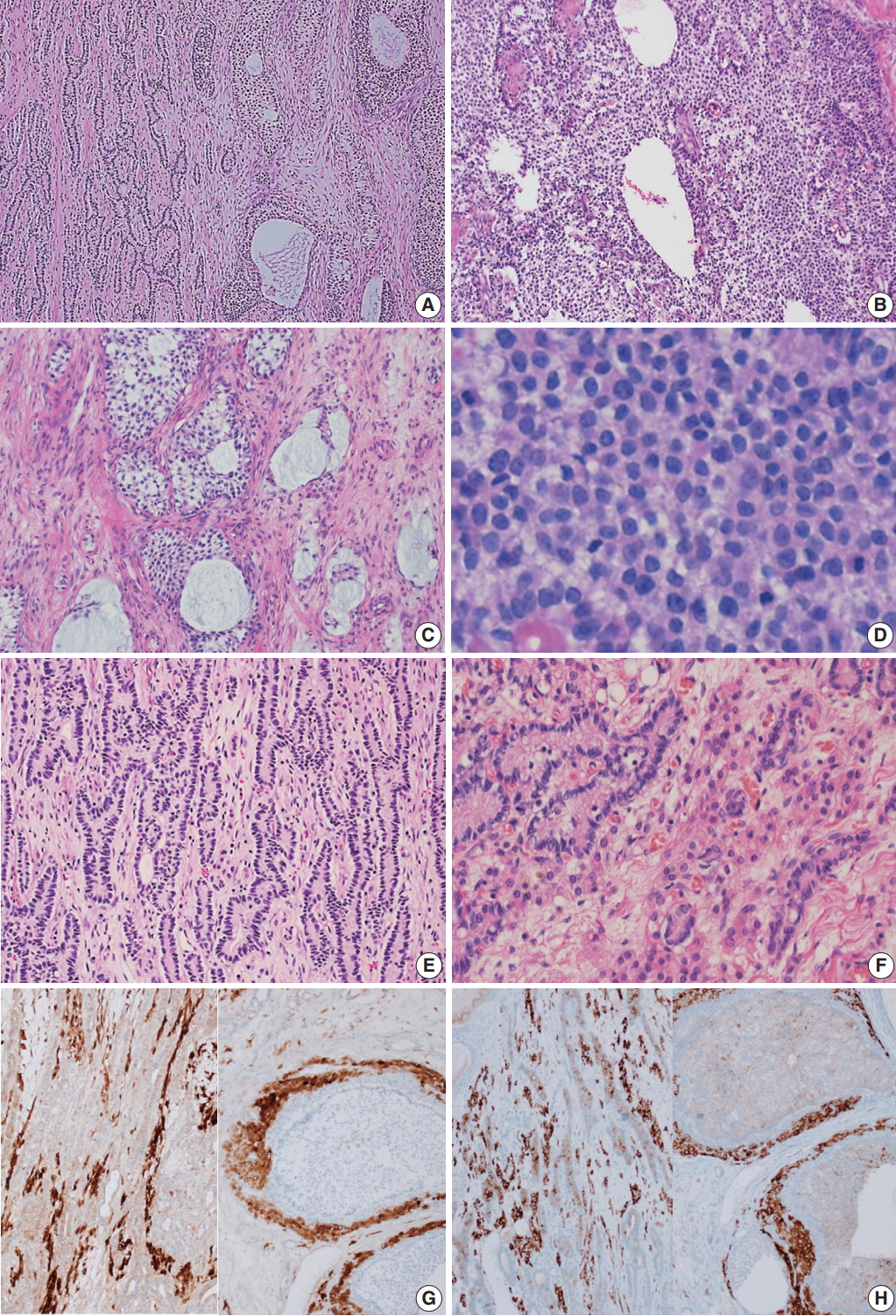
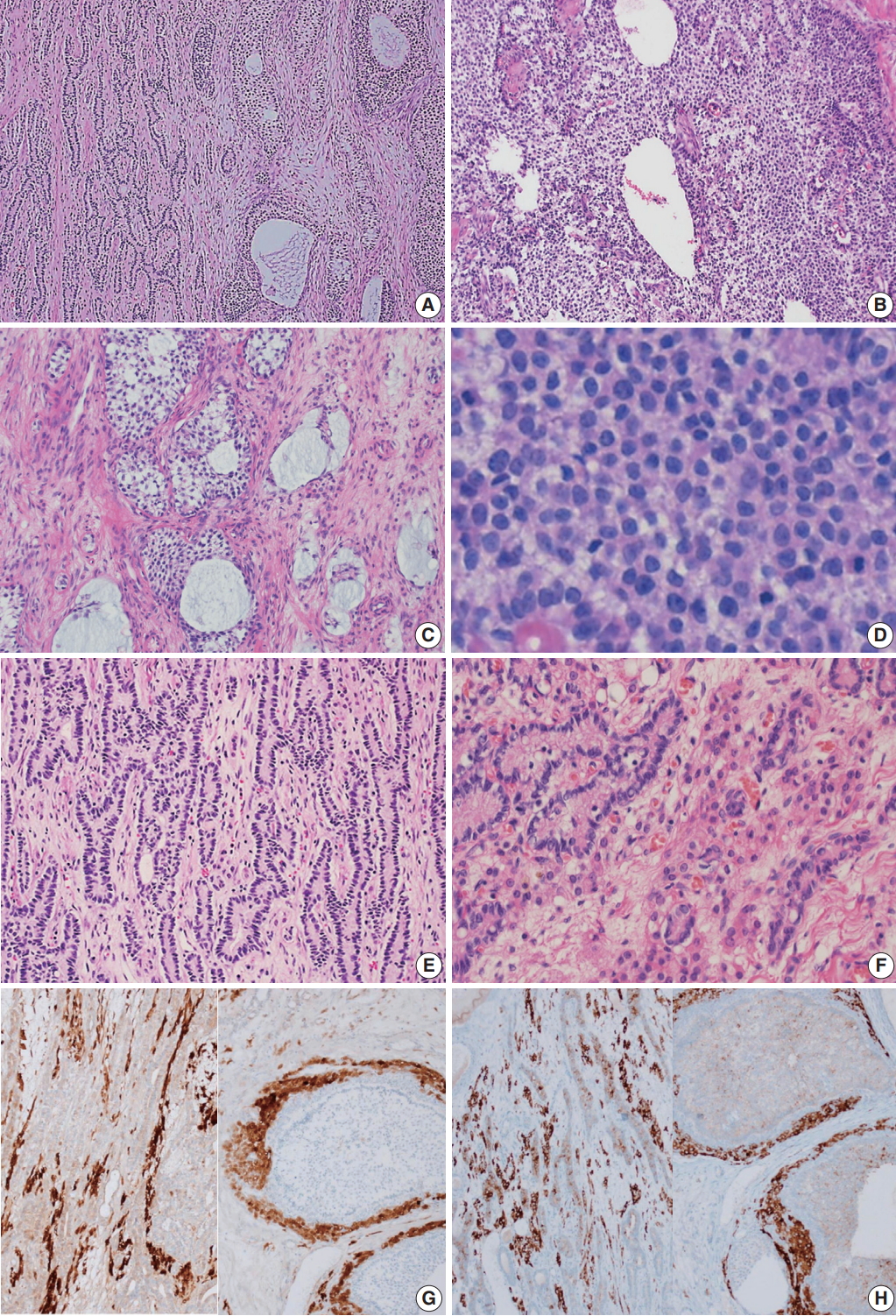
- Microscopically, the ovarian tumor consisted of a JGCT (40%) and a well-differentiated SLT (60%) (Fig. 3A). The JGCT displayed nodular growth patterns with follicles of various shapes and sizes. The follicles contained basophilic secretions resembling mucin (Fig. 3B). Cytologically, the tumor cells of the JGCT were round and had indistinct cell borders with abundant eosinophilic cytoplasm. The tumor cells’ nuclei were round-to-oval with open chromatin, but nuclear grooves were less prominent than usually observed in adult-type GCT. There was no striking nuclear atypia, and Call-Exner bodies were not identified. The SLT showed well-formed tubules of Sertoli cells and fibrous bandlike stroma with conspicuous Leydig cells (Fig. 3C). The Sertoli cells were cuboid-to-columnar with round nuclei and inconspicuous nucleoli. Nuclear atypia was absent. Leydig cells presented with abundant eosinophilic cytoplasm. Some tumor cells had foamy and vacuolated cytoplasm, but Reinke crystals were not prominent (inset of Fig. 3C).
- Immunohistochemically, the tumor cells of both tumor components were positive for calretinin and inhibin, and negative for epithelial membrane antigen (Fig. 3D–F).
- The patient had no post-surgical complications, and there was no evidence of recurrence or metastasis during two years of follow-up.
CASE REPORT
- Gynandroblastoma is a subtype of ovarian sex cord-stromal tumor and is usually observed in young women. It is identified by its admixture of ovarian and testicular tissues or cells. The most important clinical manifestation is hormonal dysfunction. Because this tumor is hormonally active, patients afflicted with it could present with either estrogenic or androgenic symptoms [4]. Gynandroblastoma with JGCT is very rare with only 6 of 28 gynandroblastoma cases reported in the English literature to date [2,3,5-8]. All reported cases occurred in premenopausal women between 15 and 39 years old. Our case is the first gynandroblastoma with JGCT diagnosed in a 71-year-old postmenopausal elderly woman. Approximately 97% of JGCTs occur during the first three decades of life. Only a few known cases of JGCT occurred in women > 30 years old; just two cases of JGCT as a component of gynandroblastoma have been reported in patients over the age of 30 [2,7]. Thus, our case of gynandroblastoma with JGCT in a postmenopausal woman is extremely unusual. However, the JGCT component of our patient’s tumor showed typical histologic features, such as variously sized follicles that contained basophilic secretions.
- The tumor diagnosis was based on histologic features. The diagnostic criteria for gynandroblastoma indicate that the minor tumor component (either Sertoli-Leydig cells in a GCT, or granulosa cells in an SLT) should account for at least 10% of the entire tumor. Sometimes, well-differentiated Sertoli-Leydig cells can be focally observed in GCTs and vice versa. Therefore, sufficient tumor sampling is essential for an accurate diagnosis. Although most reported GCT cases are of the adult-type, a juvenile-type has also been identified. The histology of adult-type and juveniletype cells is the same for GCT; therefore, it is not difficult to diagnose these tumors. Several immunohistochemical markers exist to aid differential diagnosis, but these markers are used to diagnosis every type of ovarian sex cord-stromal tumor and they are not specific to gynandroblastoma. The representative sex cord-stromal cell markers include inhibin, calretinin, SF1, WT-1, and CD99 [2]. MART-1/melan-A is a marker specific to SLT and steroid cell tumors [9]. Additionally, the cell cycle regulatory protein 14-3-3 sigma was recently identified as a diagnostic marker for GCT and steroid cell tumors [10]. In our case, both JGCT and SLT components were positive for inhibin and calretinin.
- The histogenesis of gynandroblastoma is unknown, but it is generally assumed that it originates from a single progenitor cell that can differentiate into both female and male elements [11]. During embryogenesis, gonadal tissues develop from the mesoderm of the urogenital ridge and the ridge organizes the endocrinologically active tissues of the gonad, which includes granulosa cells, Sertoli cells, theca cells, ovarian stromal cells, and Leydig cells [12]. Gynandroblastoma and other sex cord-stromal tumors are considered to have their origin in the cells of the urogenital ridge.
- It is difficult to characterize the biological behavior of gynandroblastoma due to its extremely low incidence. Based on the reported cases, it appears to have a benign course. The majority of gynandroblastomas were stage I tumors and most patients with stage I gynandroblastoma were successfully treated by simple surgical resection with regular follow-up. Only one recurrence case has been reported, in which the tumor reportedly recurred 10 years after the original tumor was surgically removed [4]. Likewise, our patient had a stage I tumor without extra-ovarian involvement. Currently, 26-month postsurgery, the patient has not shown any clinical evidence of recurrence. Given the evidence from previous reports and our current case, gynandroblastoma has a relatively benign clinical course, and our patient appears to have had a low-grade tumor.
- Recently, several genetic studies of ovarian sex cord-stromal tumors focusing on cytogenetic alterations have been published [13]. The most widely studied genetic change is the FOXL2 mutation C134W (402 C > G) found in GCTs [14]. FOXL2 is a transcription factor that is restrictedly expressed in granulosa cells during development and adulthood [15]. This mutation is widely observed in adult type-GCT and can be a diagnostic or prognostic marker [15-17]. Approximately 30% of JGCT tumors harbor the gsp oncogene and 60% contain the AKT1 gene mutation [18,19]. These biological markers have the potential to be of therapeutic value. Regarding SLT, somatic or germline mutations of DICER1, which encodes an RNase III endonuclease, are well-known genetic alterations [13]. Somatic hot spot mutations of DICER1 have been observed in approximately 60% of SLTs [20]. These frequent genetic changes of sex-cord stromal tumors had been understudied with regard gynandroblastoma. Oparka et al. [11] evaluated the C134W (402 C > G) FOXL2 mutation, a frequently observed mutation in adult-type GCT, in seven gynandroblastomas. This mutation was not detected in any GCT or SLT component of gynandroblastoma cases. However, other researchers observed FOXL2 mutations in both GCT and SLT components of two gynandroblastomas and DICER1 mutations were also observed in a few cases [13]. The therapeutic or diagnostic value of these mutations for gynandroblastoma should be studied.
- In conclusion, gynandroblastoma is an ovarian sex cord-stromal tumor with both male and female elements. Gynandroblastoma with JCGT is very rare and it typically occurs in women of reproductive age. However, it can also occur in postmenopausal women. The pathogenesis and biologic behavior of gynandroblastoma are still not well known. However, based on the limited number of previous studies, this tumor shares many clinicopathologic characteristics with other sex-cord stromal tumors, including GCT and SLT. More comprehensive histologic and genetic studies are needed.
DISCUSSION
Fig. 1.Pelvic computed tomography (CT) and ultrasonography findings. (A) Pelvic CT reveals a large multilocular cystic mass (arrow) with a solid component. (B) Ultrasonography shows a cystic mass (arrow) of approximately 13 cm arising from the right adnexa.
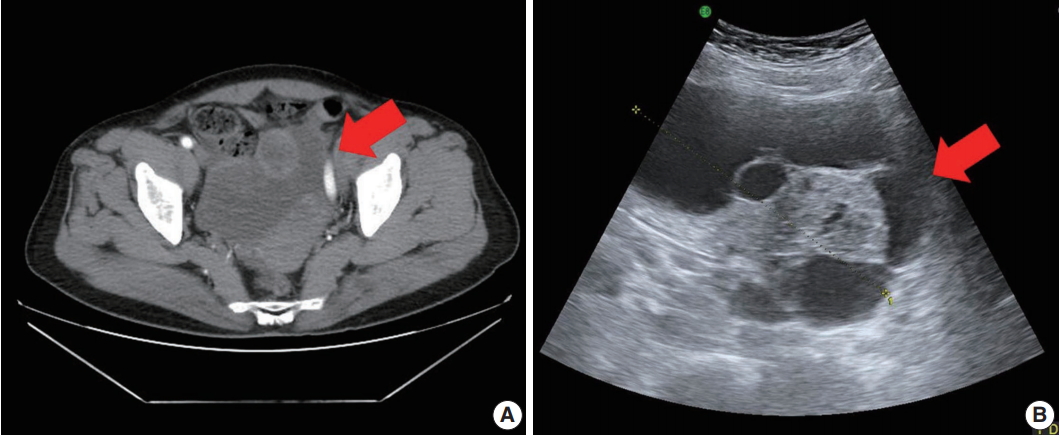
Fig. 2.Gross findings. (A) A 13.0×7.5×6.5-cm multilocular cystic and solid mass replaced the entire ovary. (B) The cysts were filled with serosanguinous fluid and approximately 30% of the mass was solid, hard, and yellow.
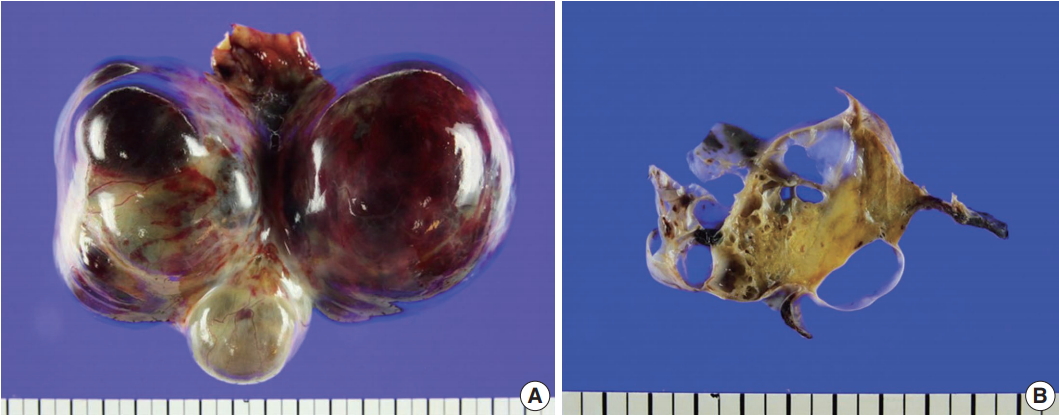
Fig. 3.Microscopic findings. (A) The tumor was composed of a Sertoli-Leydig cell tumor (SLT) and a juvenile granulosa cell tumor (JGCT). (B) The JGCT shows solid and nodular growth patterns with relatively uniform tumor cells. (C) Various sized follicles with mucin-like basophilic secretions are also frequently observed. (D) The tumor cells were rounded and had hyperchromatic nuclei without nuclear grooves, which are the characteristic features of adult granulosa cell tumors. (E) A well-differentiated SLT component is also identified. (F) Sertoli cells formed hollow tubules with delicate fibrous stroma. Small clusters of Leydig cells are observed in the fibrous stroma. (G, H) The immunohistochemical staining results confirms that the tumor was of sex cord-stromal origin. Both components stained positive for calretinin (G, left, SLT; right, JGCT) and inhibin (H, left, SLT; right, JGCT).
- 1. Mechler EA, Black WC. Gynandroblastoma of the ovary. Am J Pathol 1943; 19: 633-53. PubMedPMC
- 2. Wilberger A, Yang B. Gynandroblastoma with juvenile granulosa cell tumor and concurrent renal cell carcinoma: a case report and review of literature. Int J Surg Pathol 2015; 23: 393-8. ArticlePubMedPDF
- 3. Takeda A, Watanabe K, Hayashi S, Imoto S, Nakamura H. Gynandroblastoma with a juvenile granulosa cell component in an adolescent: case report and literature review. J Pediatr Adolesc Gynecol 2017; 30: 251-5. ArticlePubMed
- 4. Chivukula M, Hunt J, Carter G, Kelley J, Patel M, Kanbour-Shakir A. Recurrent gynandroblastoma of ovary: a case report: a molecular and immunohistochemical analysis. Int J Gynecol Pathol 2007; 26: 30-3. PubMed
- 5. Talerman A. Gynandroblastoma with elements of juvenile granulosa cell tumor. Int J Gynecol Pathol 1998; 17: 190.ArticlePubMed
- 6. Broshears JR, Roth LM. Gynandroblastoma with elements resembling juvenile granulosa cell tumor. Int J Gynecol Pathol 1997; 16: 387-91. ArticlePubMed
- 7. Chan R, Tucker M, Russell P. Ovarian gynandroblastoma with juvenile granulosa cell component and raised alpha fetoprotein. Pathology 2005; 37: 312-5. ArticlePubMed
- 8. McCluggage WG, Sloan JM, Murnaghan M, White R. Gynandroblastoma of ovary with juvenile granulosa cell component and heterologous intestinal type glands. Histopathology 1996; 29: 253-7. ArticlePubMed
- 9. Zhao C, Vinh TN, McManus K, Dabbs D, Barner R, Vang R. Identification of the most sensitive and robust immunohistochemical markers in different categories of ovarian sex cord-stromal tumors. Am J Surg Pathol 2009; 33: 354-66. ArticlePubMed
- 10. Chen L, Yang B. 14-3-3 sigma is a useful immunohistochemical marker for diagnosing ovarian granulosa cell tumors and steroid cell tumors. Int J Gynecol Pathol 2013; 32: 156-62. ArticlePubMed
- 11. Oparka R, Cassidy A, Reilly S, Stenhouse A, McCluggage WG, Herrington CS. The C134W (402 C>G) FOXL2 mutation is absent in ovarian gynandroblastoma: insights into the genesis of an unusual tumour. Histopathology 2012; 60: 838-42. ArticlePubMed
- 12. Tian W, Wang Y, Zhang H, Liu G, Ma X, Xue F. Androgen insensitivity syndrome with gynandroblastoma and vulvar leiomyoma: case report and literature review. J Low Genit Tract Dis 2013; 17: 335-9. PubMed
- 13. Conlon N, Schultheis AM, Piscuoglio S, et al. A survey of DICER1 hotspot mutations in ovarian and testicular sex cord-stromal tumors. Mod Pathol 2015; 28: 1603-12. ArticlePubMedPMCPDF
- 14. Shah SP, Kobel M, Senz J, et al. Mutation of FOXL2 in granulosacell tumors of the ovary. N Engl J Med 2009; 360: 2719-29. PubMed
- 15. D’Angelo E, Mozos A, Nakayama D, et al. Prognostic significance of FOXL2 mutation and mRNA expression in adult and juvenile granulosa cell tumors of the ovary. Mod Pathol 2011; 24: 1360-7. ArticlePubMedPDF
- 16. Jamieson S, Butzow R, Andersson N, et al. The FOXL2 C134W mutation is characteristic of adult granulosa cell tumors of the ovary. Mod Pathol 2010; 23: 1477-85. ArticlePubMedPDF
- 17. McCluggage WG, Singh N, Kommoss S, Huntsman DG, Gilks CB. Ovarian cellular fibromas lack FOXL2 mutations: a useful diagnostic adjunct in the distinction from diffuse adult granulosa cell tumor. Am J Surg Pathol 2013; 37: 1450-5. PubMed
- 18. Auguste A, Bessière L, Todeschini AL, et al. Molecular analyses of juvenile granulosa cell tumors bearing AKT1 mutations provide insights into tumor biology and therapeutic leads. Hum Mol Genet 2015; 24: 6687-98. ArticlePubMedPDF
- 19. Kalfa N, Ecochard A, Patte C, et al. Activating mutations of the stimulatory g protein in juvenile ovarian granulosa cell tumors: a new prognostic factor? J Clin Endocrinol Metab 2006; 91: 1842-7. ArticlePubMed
- 20. Heravi-Moussavi A, Anglesio MS, Cheng SW, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 2012; 366: 234-42. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Ovarian Gynandroblastoma: A Rare Sex Cord-Stromal Tumor with Unique Morphological, Genetic, and Clinical Features—A Series of Two Patients with Clinicopathological and Molecular Analysis, and a Review of the Literature
Elena Lucas, Kyle Molberg, Lesley Conrad, Shuang Niu, Hao Chen
International Journal of Surgical Pathology.2026; 34(2): 495. CrossRef - Case of Gynandroblastoma of the Ovary with Raised AFP and Associated DICER 1 Mutation
Dipak Limbachiya, Rajnish Tiwari, Rashmi Kumari, Priti Trivedi
The Journal of Obstetrics and Gynecology of India.2025; 75(S1): 549. CrossRef - Ovarian cancer in children and adolescents: A unique clinical challenge
Marina Jakimovska Stefanovska, Aleksandar Čelebić, Jean Calleja-Agius, Kristina Drusany Staric
European Journal of Surgical Oncology.2025; 51(4): 108785. CrossRef - Survival outcomes in patients with recurrent mixed sex cord-stromal tumors of the ovary
Elio Tahan, Allison L. Brodsky, Naomi R. Gonzales, Alexandra Bercow, Anil K. Sood, Lois M. Ramondetta, David M. Gershenson, R Tyler Hillman
International Journal of Gynecological Cancer.2025; 35(10): 102018. CrossRef - Rapid recurrence of ovarian mixed sex-cord-stromal tumor associated with DICER1 gene mutation: a case analysis and literature review
Fengyi Zhu, Hai Zhou, Weizheng Li
Annals of Medicine & Surgery.2025; 87(9): 6045. CrossRef - Gynandroblastoma With a Juvenile Granulosa Cell Tumor Component Presenting as Ovarian Torsion in a 14-Year-Old Female Patient
Lisa Su, Jasmine N Jefferson, Leslie Lopez-Calderon
Cureus.2025;[Epub] CrossRef - Ovarian Gynandroblastoma with a Juvenile Granulosa Cell Tumor Component in a Postmenopausal Woman
Soohyun Hwang, Byoung-Gie Kim, Sang Yong Song, Hyun-Soo Kim
Diagnostics.2020; 10(8): 537. CrossRef - Clinical and histological criteria for sex cord ovarian stromal tumors
A. М. Beishembaev, K. I. Zhordania
Obstetrics, Gynecology and Reproduction.2020; 14(3): 261. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
Ovarian Gynandroblastoma with a Juvenile Granulosa Cell Tumor Component in a Postmenopausal Woman: A Case Report and Literature Review



Fig. 1. Pelvic computed tomography (CT) and ultrasonography findings. (A) Pelvic CT reveals a large multilocular cystic mass (arrow) with a solid component. (B) Ultrasonography shows a cystic mass (arrow) of approximately 13 cm arising from the right adnexa.
Fig. 2. Gross findings. (A) A 13.0×7.5×6.5-cm multilocular cystic and solid mass replaced the entire ovary. (B) The cysts were filled with serosanguinous fluid and approximately 30% of the mass was solid, hard, and yellow.
Fig. 3. Microscopic findings. (A) The tumor was composed of a Sertoli-Leydig cell tumor (SLT) and a juvenile granulosa cell tumor (JGCT). (B) The JGCT shows solid and nodular growth patterns with relatively uniform tumor cells. (C) Various sized follicles with mucin-like basophilic secretions are also frequently observed. (D) The tumor cells were rounded and had hyperchromatic nuclei without nuclear grooves, which are the characteristic features of adult granulosa cell tumors. (E) A well-differentiated SLT component is also identified. (F) Sertoli cells formed hollow tubules with delicate fibrous stroma. Small clusters of Leydig cells are observed in the fibrous stroma. (G, H) The immunohistochemical staining results confirms that the tumor was of sex cord-stromal origin. Both components stained positive for calretinin (G, left, SLT; right, JGCT) and inhibin (H, left, SLT; right, JGCT).
Fig. 1.
Fig. 2.
Fig. 3.
Ovarian Gynandroblastoma with a Juvenile Granulosa Cell Tumor Component in a Postmenopausal Woman: A Case Report and Literature Review

