 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 57(4); 2023 > Article
-
Review
A review of liver fibrosis and cirrhosis regression -
Michael J. Lee

-
Journal of Pathology and Translational Medicine 2023;57(4):189-195.
DOI: https://doi.org/10.4132/jptm.2023.05.24
Published online: June 20, 2023
Department of Pathology and Cell Biology, Columbia University Medical Center, New York, NY, USA
- Corresponding Author: Michael J. Lee, MD, Department of Pathology and Cell Biology, Columbia University Medical Center, 630 West 168th Street, VC14-240A, New York, NY 10032, USA Tel: +1-212-342-3016, E-mail: mjl2197@cumc.columbia.edu
© 2023The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Cirrhosis has traditionally been considered an irreversible process of end-stage liver disease. With new treatments for chronic liver disease, there is regression of fibrosis and cirrhosis, improvement in clinical parameters (i.e. liver function and hemodynamic markers, hepatic venous pressure gradient), and survival rates, demonstrating that fibrosis and fibrolysis are a dynamic process moving in two directions. Microscopically, hepatocytes push into thinning fibrous septa with eventual perforation leaving behind delicate periportal spikes in the portal tracts and loss of portal veins. Obliterated portal veins during progressive fibrosis and cirrhosis due to parenchymal extinction, vascular remodeling and thrombosis often leave behind a bile duct and hepatic artery within the portal tract. Traditional staging classification systems focused on a linear, progressive process; however, the Beijing classification system incorporates both the bidirectional nature for the progression and regression of fibrosis. However, even with regression, vascular lesions/remodeling, parenchymal extinction and a cumulative mutational burden place patients at an increased risk for developing hepatocellular carcinoma and should continue to undergo active clinical surveillance. It is more appropriate to consider cirrhosis as another stage in the evolution of chronic liver disease as a bidirectional process rather than an end-stage, irreversible state.
- With chronic and persistent liver injury, the liver responds by depositing extracellular matrix (ECM). This is characterized by type 1 and 3 collagen deposition in the portal tracts and lobules, and collagenous and non-collagenous ECM protein deposition in the space of Disse, which includes collagen type 3 and 4, laminin and fibronectin [8]. This wound healing response or scarring down of the liver begins when chronic liver injury causes apoptosis of hepatocytes leading to Kupffer cell activation and cytokine release: tumor necrosis factor (TNF), platelet derived growth factor (PDGF), and endothelin-1 (ET-1). Quiescent hepatic stellate cells (HSCs) in the space of Disse are normally inactive, dormant, fat storing cells, however, TNF activates stellate cells for transformation into myofibroblast like cells, depositing ECM as collagen types 1, 3, 4, and laminin. PDGF causes proliferation of stellate cells and ET-1 leads to contraction of stellate cells and vasoconstriction, affecting vascular resistance and liver blood flow. While normal sinusoidal spaces are lined by fenestrated endothelial cells, increased ECM material and collagen deposition closes sinusoidal endothelial cell fenestrations and the space of Disse, a process called capillarization of the sinusoids, preventing protein exchange between hepatocytes and flowing plasma [8-10].
- Chronic portal and lobular inflammation causing hepatocellular necrosis with subsequent parenchymal extinction not only represents hepatocellular loss but an alteration in the surrounding microvasculature as chronic inflammation causes thrombosis of small branches of the portal vein, hepatic vein, and hepatic artery. This ischemic injury leads to subsequent cycles of parenchymal loss with compounding vascular compromise and remodeling, bile ductular reaction, and collapse of the hepatocellular trabeculae. When chronic injury persists, there can be years of parenchymal, architectural, and vascular remodeling, the latter which may be irreversible [2,11,12].
- In contrast, matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs) are enzymes responsible for breaking down the extracellular matrix during fibrosis regression. There are approximately nine MMPs involved in the reversal of fibrosis with varying substrate specificity. These proteases are subcategorized into the following groups: stromelysins that break down protein substrates (i.e., laminin, gelatin, and type IV collagen), collagenases that degrade collagen (i.e., type III and I collagen), and gelatinases/type IV collagenases. MMPs are balanced and regulated by TIMPs, which irreversibly bind to and inhibit MMPs and are released by HSCs and Kupffer cells [13-15].
- Retinoids (vitamin A) keep stellate cells in the quiescent state and prevent conversion to an activated state, inhibiting further differentiation into a myofibroblast-like phenotype. In tissue culture, activated HSCs show loss of retinoids, however, adding retinoids to the culture medium decreased type 1 collagen synthesis and deposition. Experimental studies showed that exogenous retinoid administration reduced expression of inflammatory cytokines such as TNF-α, maintained HSCs in a quiescent state and prevented Kupffer cells from releasing fibrogenic cytokines. Thus, there is an ongoing balance between fibrogenesis and fibrolysis involving an interplay of numerous factors during this dynamic, bidirectional process [16,17].
PATHOBIOLOGY
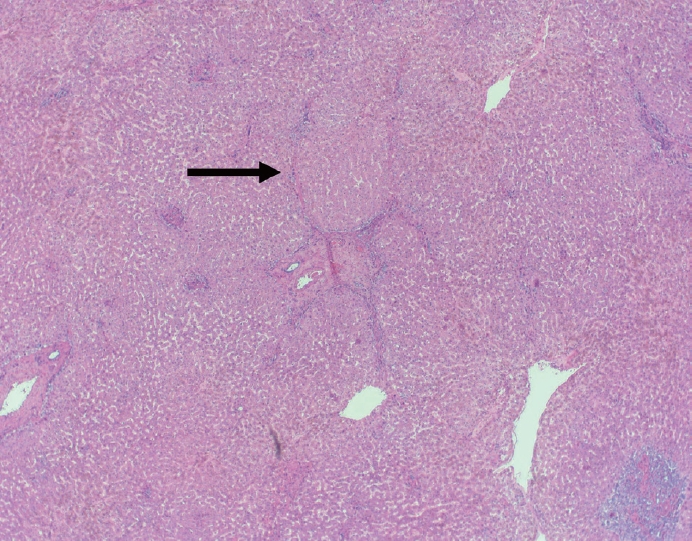
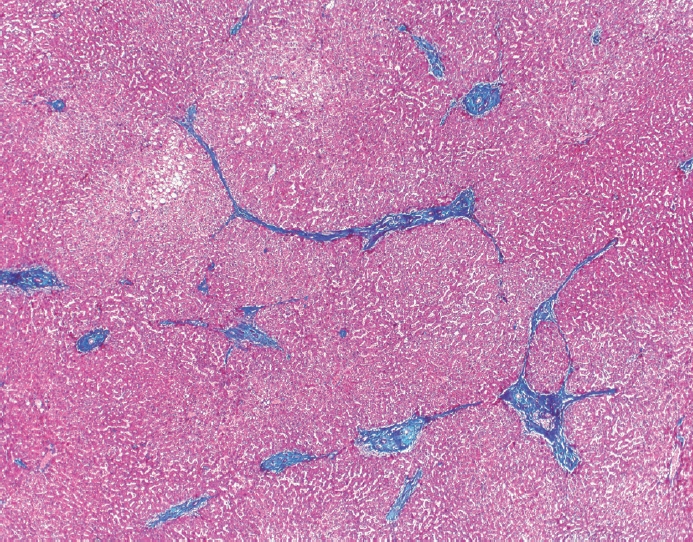
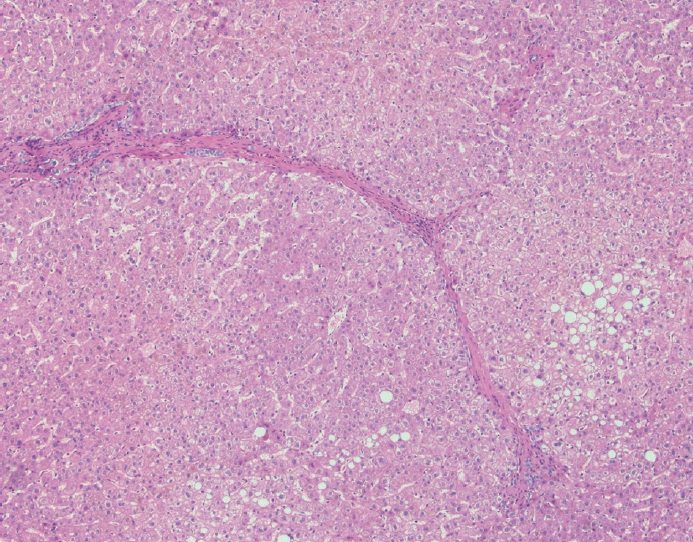
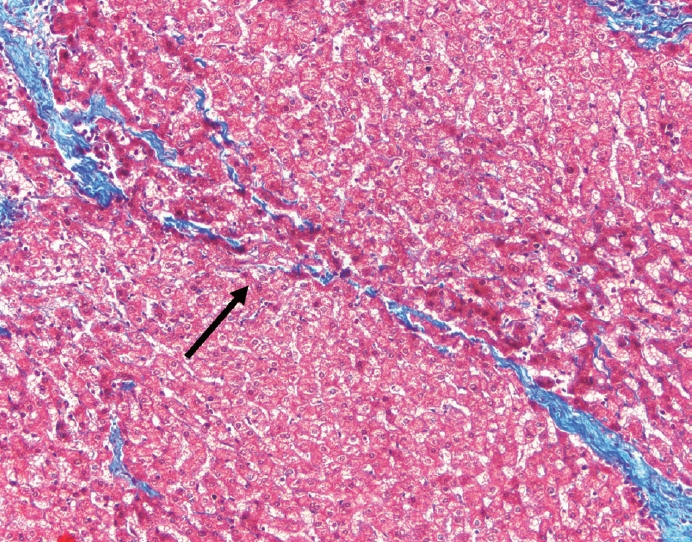
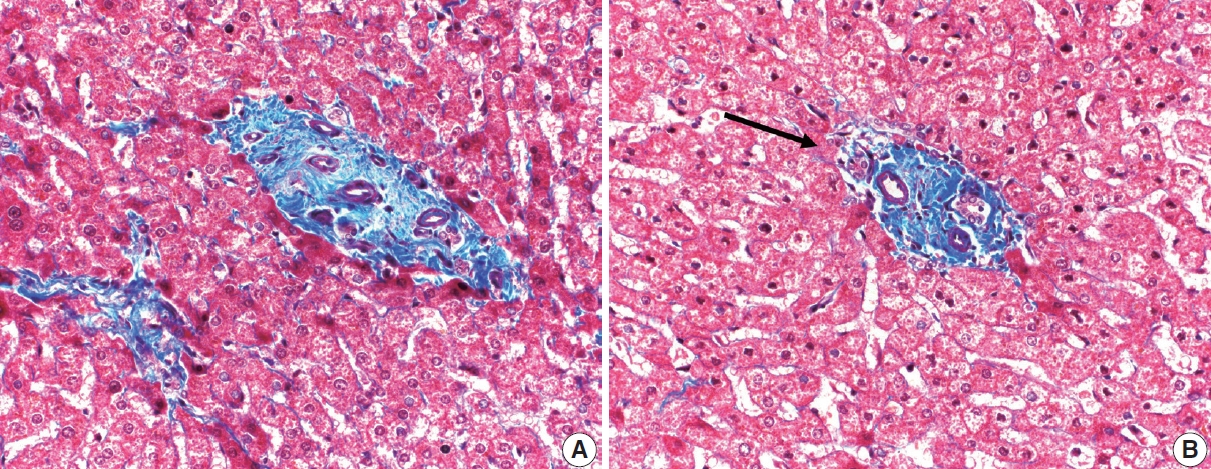
- The typical cirrhotic nodule is surrounded by broad, thick fibrous septae with scattered chronic inflammation and bile ductular reaction encapsulating a nodule of regenerating hepatocytes. The central veins may not be visualized because they have been compressed and undergo thrombosis. As these outflow vessels are obliterated, small, collateral vascular channels develop and act as shunts to offset the increase in portal pressure. In contrast, fibrosis regression is comprised of three components, fragmentation and regression of the scar, vascular remodeling/distortion, and parenchymal regeneration. Inflammation subsides as the balance is shifted towards fibrolysis and fibrous septa become progressively thinner and wispier (Figs. 1–3). Eventually, hepatocytes push into or split the fibrous septa causing small perforations and fragmenting the septa (Figs. 4, 5). As the liver reverses bridging fibrosis and cirrhosis, delicate periportal fibrous spikes are left behind in the portal tracts [18,19].
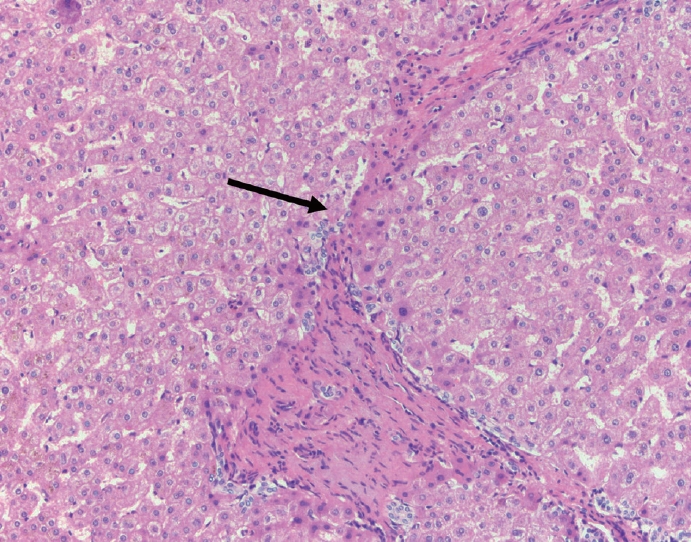
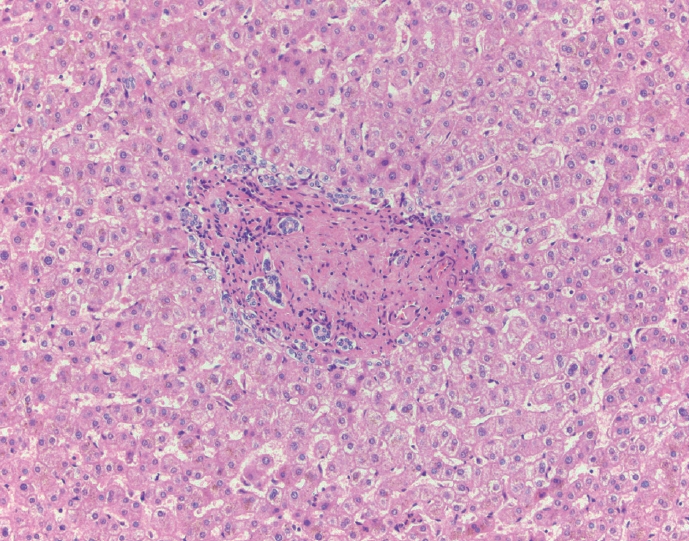
- The portal tracts also demonstrate fibrosis regression as collagen is broken down, often leaving behind a paired bile duct and hepatic artery while the previously obliterated portal vein is not restored or visualized. This is known as a portal tract remnant. Hepatocytes prolapse or push into the fibroconnective tissue boundaries of the portal tract and are seen directly adjacent to the paired bile duct and artery (Figs. 6, 7). As the inflammation and hepatocellular extinction subsides, parenchymal regeneration begins, sinusoidal collagen is resorbed and hepatic trabeculae restores its normal architecture [18,20,21].
PATHOLOGIC FEATURES
- Traditional classification staging systems (i.e., Batts-Ludwig, Ishak, Metavir) characterized the architectural changes of fibrosis and cirrhosis in a linear pattern from no fibrosis to portal expansion, followed by step-wise periportal fibrosis, bridging fibrosis and ultimately cirrhosis (Table 1) [22,23]. Kutami et al. [24] introduced the Laennec classification system in 2000, which subdivided stage 4 cirrhosis into three categories: 4A, 4B, and 4C. Stage 4A was deemed mild cirrhosis with thin fibrous septa and large nodules, 4B with at least two broad septa but small nodules and stage 4C with large, broad fibrous septa with several small nodules. While this system is easily reproducible, there has not been widespread consensus in adopting this classification scheme [24].
- Due to effective treatments for chronic liver disease and the consideration of fibrosis and cirrhosis regression as a dynamic, bidirectional two-way street, Thiese et al. [18] proposed a new classification system for grading and staging hepatitis patients, the Beijing classification (Table 2). This system was proposed for the assessment of chronic viral hepatitis but has proven useful in describing activity and fibrosis for other etiologies of chronic hepatitis. It simplified grading activity and staging fibrosis and added a new, third category for determining the quality of fibrosis. Each biopsy is assessed a P-I-R score, predominantly progressive, indeterminate, or predominantly regressive as the major pattern of fibrosis. Predominantly progressive features show broad, thick fibrous septa with chronic inflammation, ductular reaction, parenchymal extinction, and congestion. A regressive pattern demonstrates wispy, thin fibrous septa with fragmentation, perforation, little or no inflammation and prolapsing hepatocytes into the portal tract as described earlier. Indeterminate denotes that the surgical pathologist is unable to distinctly classify a biopsy as P or R. The main benefit is that post-treatment biopsies can remain in the same stage (i.e., traditional stage 4 cirrhosis), but show significant changes in fibrosis quality (i.e., progressive to regressive or indeterminate), providing valuable prognostic information and determining response to therapy [18].
- Besides a P-I-R score, liver biopsies are also graded and staged in the Beijing classification system (Table 2). Necroinflammation or hepatitis is graded as inactive (only portal inflammation or rare interface or lobular activity), non-severe active (variable interface and lobular hepatitis), and severe active (confluent necrosis, perivenular or bridging necrosis). Fibrosis is staged as early (no fibrosis, portal fibrosis), intermediate (focal or frequent fibrous septa, bridging fibrosis) and advanced (fibrous septa with focal or diffuse nodularity) [18]. The similarities to prior adopted grading and staging classification systems are evident. This new system for microscopic evaluation of grading and staging is susceptible to intra and interobserver variability, therefore, simplifying and decreasing the number of subcategories in grading activity and staging fibrosis improves reproducibility. Interobserver agreement between pathologists adopting the P-I-R staging system was high with a Kappa value of 0.71 (substantial agreement) [18,25]. The P-I-R system is also a valuable prognostic marker independent of the grade and stage, providing a snapshot for the current state of disease that strongly correlates with hepatic venous wedge pressures (hepatic venous pressure gradient [HVPG]) and portal hypertension [26]. In patients with chronic viral hepatitis, P, I, or R was an accurate surrogate marker for clinical outcome as successful eradication and clearance of hepatotropic viruses were predominantly R and unsuccessful treatments were predominantly P or I [18].
CLASSIFICATION SYSTEMS
- In advanced liver disease, there is increased resistance to sinusoidal blood flow which causes portal hypertension or increased portal pressures. The gold standard for measuring portal hypertension is the HVPG and a gradient of less than or equal to 5 is within normal limits. Cirrhotic patients vary widely in their clinical presentation because the severity of cirrhosis ranges from compensated and asymptomatic to decompensated cirrhosis with ascites, esophageal varices, and hepatic encephalopathy. HVPG is an accurate prognostic marker in cirrhotic patients that risk stratifies the likelihood of those complications. Clinical studies show that with cirrhosis and fibrosis regression, there is a decrease in HVPG and portal hypertension-related complications with improvements in liver function and survival rates. A comprehensive assessment is based on clinical, hemodynamic (i.e., HVPG), and histopathologic features [27-29].
- There are multiple approaches to achieving reversal of fibrosis and cirrhosis. The most common method is to control or cure the primary, underlying disease. This has a proven and successful track record for hepatotropic viruses (i.e., hepatitis B and C), autoimmune hepatitis, hereditary hemochromatosis, Wilson’s disease, and fatty liver disease. Anti-fibrotic agents target different steps in the pathobiology of fibrosis. These therapeutic drugs are focused on receptor-ligand interactions to prevent quiescent HSCs from transforming into activated HSCs, preventing the cascade of events that lead to deposition of ECM by inhibiting fibrogenesis, or accentuating the resolution of fibrosis through apoptosis or increased matrix degradation. There are over 500 active, clinical trials in this area of research [30].
- Cirrhosis is also a major risk factor for developing hepatocellular carcinoma (HCC). With regression, there are physiologic and mechanical/pressure improvements in the patient’s condition, however, is there a reduction in the risk for developing HCC? There are two main factors that contribute to the pathobiology of HCC, the cumulative mutations of liver disease etiology (i.e., viral, metabolic, fatty liver, etc.) that form a clonal population/neoplasm and the surrounding extracellular matrix/ tumor microenvironment (TME) which consists of vascular abnormalities/remodeling and the fibrous stroma [31- 33]. Even when the initial insult is removed (i.e., hepatotropic virus cleared by medication, weight loss or medication for steatohepatitis, phlebotomy for hemochromatosis, etc.) and fibrosis regression is visualized, the driver mutations within hepatocytes persist. For example, in hepatitis B, there is DNA integration into the host genome leading to genomic instability, alterations to tumor suppressor genes, and TP53 mutations [34,35]. In non-alcoholic fatty liver disease, there are a different set of cumulative mutations, alterations in fatty acid beta-oxidation and insulin resistance [36]. As for the TME, vascular remodeling, ischemic injury, thrombosis of small blood vessels, lead to cycles of parenchymal extinction and regeneration with increased vascular endothelial growth factor expression and further genomic instability. Therefore, despite fibrosis regression, the risk for developing HCC remains high compared to the normal population and patients should continue to be actively screened and followed [32].
CLINICAL IMPLICATIONS
- Cirrhosis was once thought to be an irreversible process of endstage liver disease. This is no longer the case with treatment options for most chronic liver diseases. Fibrosis regression is characterized by thinning of the fibrous septa with hepatocytes pushing into the septa and eventual perforation. This leads to periportal spiking within portal tracts and prolapsed hepatocytes into the boundaries of the portal tract stroma. Obliterated portal veins during progressive fibrosis and cirrhosis due to parenchymal extinction, vascular remodeling, and thrombosis often leaves behind a bile duct and hepatic artery within the portal tract. To characterize these histopathologic features, the Beijing classification system offers an accurate snapshot of a dynamic process between fibrogenesis and fibrolysis. Even with fibrosis regression, vascular lesions/remodeling, parenchymal extinction, and cumulative mutational burden persists and patients should continue to undergo clinical surveillance.
CONCLUSION
Ethics Statement
Not applicable.
Data availability
All data generated or analyzed during the study are included in this published article (and its supplementary information files).
Code Availability
Not applicable.
Conflicts of Interest
The authors declare that they have no potential conflicts of interest.
Funding Statement
No funding to declare.
- 1. MacSween RN, Burt AD, Portman B, et al. Pathology of the liver. 4th ed. Philadelphia: Saunders, 2002.
- 2. Wanless IR, Nakashima E, Sherman M. Regression of human cirrhosis: morphologic features and the genesis of incomplete septal cirrhosis. Arch Pathol Lab Med 2000; 124: 1599-607. PubMed
- 3. Hutterer F, Rubin E, Popper H. Mechanism of collagen resorption in reversible hepatic fibrosis. Exp Mol Pathol 1964; 3: 215-23. ArticlePubMed
- 4. Brody JI, Mc KD, Kimball SG. Therapeutic phlebotomies in idiopathic hemochromatosis. Am J Med Sci 1962; 244: 575-86. PubMed
- 5. Perez-Tamayo R. Cirrhosis of the liver: a reversible disease? Pathol Annu 1979; 14 Pt 2: 183-213. PubMed
- 6. Geller SA. Coming or going? What is cirrhosis? Arch Pathol Lab Med 2000; 124: 1587-8. ArticlePubMedPDF
- 7. Chejfec G. Controversies in pathology: is cirrhosis of the liver a reversible disease? Arch Pathol Lab Med 2000; 124: 1585-6. PubMed
- 8. Martinez-Hernandez A, Martinez J. The role of capillarization in hepatic failure: studies in carbon tetrachloride-induced cirrhosis. Hepatology 1991; 14: 864-74. ArticlePubMed
- 9. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008; 134: 1655-69. ArticlePubMed
- 10. Mori T, Okanoue T, Sawa Y, Hori N, Ohta M, Kagawa K. Defenestration of the sinusoidal endothelial cell in a rat model of cirrhosis. Hepatology 1993; 17: 891-7. ArticlePubMed
- 11. Guido M, Sarcognato S, Russo FP, et al. Focus on histological abnormalities of intrahepatic vasculature in chronic viral hepatitis. Liver Int 2018; 38: 1770-6. ArticlePubMedPDF
- 12. Schaffner F, Poper H. Capillarization of hepatic sinusoids in man. Gastroenterology 1963; 44: 239-42. ArticlePubMed
- 13. Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis 2001; 21: 373-84. ArticlePubMed
- 14. de Meijer VE, Sverdlov DY, Popov Y, et al. Broad-spectrum matrix metalloproteinase inhibition curbs inflammation and liver injury but aggravates experimental liver fibrosis in mice. PLoS One 2010; 5: e11256. ArticlePubMedPMC
- 15. Muller D, Quantin B, Gesnel MC, Millon-Collard R, Abecassis J, Breathnach R. The collagenase gene family in humans consists of at least four members. Biochem J 1988; 253: 187-92. ArticlePubMedPMCPDF
- 16. Yoneda A, Sakai-Sawada K, Niitsu Y, Tamura Y. Vitamin A and insulin are required for the maintenance of hepatic stellate cell quiescence. Exp Cell Res 2016; 341: 8-17. ArticlePubMed
- 17. Davis BH, Pratt BM, Madri JA. Retinol and extracellular collagen matrices modulate hepatic Ito cell collagen phenotype and cellular retinol binding protein levels. J Biol Chem 1987; 262: 10280-6. ArticlePubMed
- 18. Theise ND, Jia J, Sun Y, Wee A, You H. Progression and regression of fibrosis in viral hepatitis in the treatment era: the Beijing classification. Mod Pathol 2018; 31: 1191-200. ArticlePubMedPDF
- 19. Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology 1995; 21: 1238-47. ArticlePubMed
- 20. Taguchi K, Asano G. Neovascularization of pericellular fibrosis in alcoholic liver disease. Acta Pathol Jpn 1988; 38: 615-26. ArticlePubMed
- 21. Wanless IR. The role of vascular injury and congestion in the pathogenesis of cirrhosis: the congestive escalator and the parenchymal extinction sequence. Curr Hepatol Rep 2020; 19: 40-53. ArticlePDF
- 22. Batts KP, Ludwig J. Chronic hepatitis: an update on terminology and reporting. Am J Surg Pathol 1995; 19: 1409-17. PubMed
- 23. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology 1996; 24: 289-93. ArticlePubMed
- 24. Kutami R, Girgrah N, Wanless I, et al. The Laennec grading system for assessment of hepatic fibrosis: validation by correlation with wedged hepatic vein pressure and clinical features. Hepatology 2000; 32: 407.
- 25. Sun Y, Zhou J, Wang L, et al. New classification of liver biopsy assessment for fibrosis in chronic hepatitis B patients before and after treatment. Hepatology 2017; 65: 1438-50. ArticlePubMedPDF
- 26. Garcia-Tsao G, Friedman S, Iredale J, Pinzani M. Now there are many (stages) where before there was one: in search of a pathophysiological classification of cirrhosis. Hepatology 2010; 51: 1445-9. PubMed
- 27. Bochnakova T. Hepatic venous pressure gradient. Clin Liver Dis (Hoboken) 2021; 17: 144-8. ArticlePubMedPMCPDF
- 28. Mauro E, Crespo G, Montironi C, et al. Portal pressure and liver stiffness measurements in the prediction of fibrosis regression after sustained virological response in recurrent hepatitis C. Hepatology 2018; 67: 1683-94. ArticlePubMedPDF
- 29. Sanyal AJ, Anstee QM, Trauner M, et al. Cirrhosis regression is associated with improved clinical outcomes in patients with nonalcoholic steatohepatitis. Hepatology 2022; 75: 1235-46. ArticlePubMedPDF
- 30. Yoon YJ, Friedman SL, Lee YA. Antifibrotic therapies: where are we now? Semin Liver Dis 2016; 36: 87-98. ArticlePubMed
- 31. Santhakumar C, Gane EJ, Liu K, McCaughan GW. Current perspectives on the tumor microenvironment in hepatocellular carcinoma. Hepatol Int 2020; 14: 947-57. ArticlePubMedPDF
- 32. Lin CA, Chang LL, Zhu H, He QJ, Yang B. Hypoxic microenvironment and hepatocellular carcinoma treatment. Hepatoma Res 2018; 4: 26.Article
- 33. Capece D, Fischietti M, Verzella D, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int 2013; 2013: 187204.ArticlePubMedPDF
- 34. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol 2016; 64(1 Suppl): S84-101. ArticlePubMed
- 35. Niu B, Hann HW. Hepatitis B virus–related hepatocellular carcinoma: carcinogenesis, prevention, and treatment. In: Abdeldayem HM, ed. Updates in liver cancer. London: IntechOpen Ltd, 2017.
- 36. Peiseler M, Tacke F. Inflammatory mechanisms underlying nonalcoholic steatohepatitis and the transition to hepatocellular carcinoma. Cancers (Basel) 2021; 13: 730.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Ultrasound‐Responsive Dual‐Prodrug Nanoassembly for “Fenestrae‐Restoration Strategy” in Liver Fibrosis Therapy
Shutong Liu, Mengyao Zhang, Zitong Qiu, Xinyi Liu, Yunsheng Dou, Sona Krizkova, Zheyi Li, Zbynek Heger, Nan Li
Advanced Materials.2026;[Epub] CrossRef - Molecular and cellular secrets revealed: How umbilical cord-derived mesenchymal stem cells can target hepatocellular carcinoma
Mohammad Sadegh Izadi, Reza Arefnezhad, Amirmasoud Asadi, Aryan Rezaee, Leila Kalantari, Farzad Nasrpour Tahouneh, Mohammad Mehdi Shadravan, Seyyed Taher Seyyed Mahmoudi, Zahra GhaniBeygi, Fatemeh Rostamnezhad, Armin Razman, Fardad Ejtehadi, Fatemeh Rezae
Tissue and Cell.2026; 98: 103200. CrossRef - Diagnostic Utility of Apparent Diffusion Coefficient Values of Spleen and Liver in Assessment of Severity of Portal Hypertension and Liver Cirrhosis
Kommuri Siddhardha, S. Jalaludheen, Senthil Kumar Aiyappan
Nigerian Postgraduate Medical Journal.2026; 33(1): 118. CrossRef - Head to head: should portal inflammation be part of grading necroinflammatory activity in metabolic dysfunction‐associated steatotic liver disease?
Daniela Allende, Alastair D Burt
Histopathology.2026; 88(5): 958. CrossRef - Diagnostic accuracy of deep learning-enhanced MRI techniques for liver fibrosis and cirrhosis detection: a systematic review and meta-analysis
Mohammadreza Elhaie, Abolfazl Koozari, Iraj Abedi
Egyptian Liver Journal.2026;[Epub] CrossRef - Physical Activity and Liver Fibrosis: A Stratified Analysis by Obesity and Diabetes Status
Junghwan Cho, Sunghwan Suh, Ji Min Han, Hye In Kim, Hanaro Park, Hye Rang Bak, Ji Cheol Bae
Journal of Clinical Medicine.2026; 15(2): 757. CrossRef - Serum Kallistatin level as a biomarker for evaluation of liver function status in cirrhotic patients
Mahmoud Rizk, F. M. Khalil, Mohamed El-Assal, Hussien Ali, Ahmed R. Mohamed, Samer S. Zekry, Abeer Mahmoud El-Bahy, Amira Kamal ElDin Mohammed ElAlfy
Egyptian Liver Journal.2026;[Epub] CrossRef - The Dual-Faceted Role of Metal-Based Nanomaterials in Hepatic Fibrosis Therapy
Yinqing Mao, Yankai Gong, Xue Bai
International Journal of Nanomedicine.2026; Volume 21: 1. CrossRef - Reply to the Letter on “Clinical implications of fibrosis marker dynamics after hepatitis C cure: insights from paired biopsies”
Hung-Wei Wang, Cheng-Yuan Peng
Journal of the Formosan Medical Association.2026;[Epub] CrossRef - Risk factors for advanced liver fibrosis among people living with HIV: a 5-year longitudinal study in Thailand
Thanawat Nochaiwong, Jutatip Sillabutra, Chukiat Viwatwongkasem, Chuleeporn Wongvoranet, Hay Mar Su Lwin, Napon Hiranburana, Anchalee Avihingsanon
AIDS Care.2026; : 1. CrossRef - The role and possible mechanism of intestinal fungi in the progression of chronic liver diseases
Yirui Hu, Ye Yang, Shuyan Wang, Huikuan Chu
npj Biofilms and Microbiomes.2026;[Epub] CrossRef - Ishak stage 6 fibrosis is more likely to regress than stage 5 in CHB patients undergoing entecavir therapy
Mengjie Li, Xiaogang Hao, Yuanyuan Li, Chao Zhang, Yongping Chen, Zujiang Yu, Qin Li, Lin Tan, Dedong Xiang, Qinghua Shang, Chunliang Lei, Liang Chen, Xiaoyu Hu, Jing Wang, Huabao Liu, Wei Lu, Yan Chen, Zheng Dong, Wenlin Bai, Eric M. Yoshida, Nahum Mende
Frontiers in Medicine.2026;[Epub] CrossRef - ECM1 produced by hepatic stellate cells serves as a gatekeeper of liver homeostasis in hepatic fibrosis
Aiting Yang, Xuzhen Yan, Yiwen Wang, Qi Han, Xiaofei Tong, Shuyan Chen, Xinyu Zhao, Wei Chen, Jidong Jia, Detlef Schuppan, Hong You
Hepatology.2026;[Epub] CrossRef - Silent progression and therapeutic reversibility of pediatric hepatitis B virus-related cirrhosis: Needs unmet
Larry Huang, Jialing Huang
World Journal of Gastroenterology.2026;[Epub] CrossRef - Double-target responsive FRET sensor based on DNA tetrahedron for robust detection of liver fibrosis-related mRNAs in activated hepatic stellate cells
Jinyong Wang, Jiayin Huang, Meiqin Zhang, Xinyao Liu, Yanlin Liu, Ya Zhang, Xiaodong Wang, Hongfei Zhou, Dazhi Chen, Zhifa Shen, Yongping Chen
Biosensors and Bioelectronics.2026; 307: 118737. CrossRef - Extracellular Vesicles in Hepatic Pathogenesis: From Biogenesis to Biomarkers
Tamer A. Addissouky
Archives of Molecular Biology and Genetics.2026; 5(1): 10. CrossRef - Evaluation of Hepatic Enzymes and Their Association with Disease Severity in Liver Cirrhosis Patients
Saif M. Hasan, Nabaa Qassim Saad-Allah, Ibrahim Khalaf Hameed
Journal of Clinical Practice and Medical Research.2026; 2(3): 57. CrossRef - A Delphi Study on the Development of a Traditional Chinese Medicine Syndrome–Differentiation Nursing Framework for Patients with Cirrhotic Ascites
Suling Huang, Linmei Zhang, Sun Yang, Fei Zheng, Sheng Tan, Danping Huang
Journal of Multidisciplinary Healthcare.2026; Volume 19: 1. CrossRef - Human donor liver viability evaluation with polarization-sensitive optical coherence tomography
Feng Yan, Qinghao Zhang, Bornface M. Mutembei, Ke Zhang, Yan Cui, Ronghao Liu, Junyuan Liu, Chen Wang, Zaid A. Alhajeri, Kaustubh Pandit, Fan Zhang, Zhongxin Yu, Kar-Ming Fung, Shaima N. Elgenaid, Paige Parrack, Walid Ali, Clint A. Hostetler, Ashley N. Mi
Science Translational Medicine.2026;[Epub] CrossRef - Regression of liver cirrhosis
Jonathan Andrew Fallowfield, Prakash Ramachandran, Timothy James Kendall
Journal of Hepatology.2026;[Epub] CrossRef - Low-Grade Chronic Inflammation: a Shared Mechanism for Chronic Diseases
Mariana Cifuentes, Hugo E. Verdejo, Pablo F. Castro, Alejandro H. Corvalan, Catterina Ferreccio, Andrew F. G. Quest, Marcelo J. Kogan, Sergio Lavandero
Physiology.2025; 40(1): 4. CrossRef - Natural History of Metabolic Dysfunction-Associated Steatotic Liver Disease: From Metabolic Syndrome to Hepatocellular Carcinoma
Melchor Alpízar Salazar, Samantha Estefanía Olguín Reyes, Andrea Medina Estévez, Julieta Alejandra Saturno Lobos, Jesús Manuel De Aldecoa Castillo, Juan Carlos Carrera Aguas, Samary Alaniz Monreal, José Antonio Navarro Rodríguez, Dulce María Fernanda Alpí
Medicina.2025; 61(1): 88. CrossRef - Unveiling the Link between Albumin-Bilirubin Grade and Liver Fibrosis in Patients with a History of Gallstone and Gallbladder Surgery: A Focus on Metabolic Dysfunction-Associated Steatohepatitis
Mohammadjavad Sotoudeheian
The Korean Journal of Pancreas and Biliary Tract.2025; 30(1): 10. CrossRef - Impact of Weight Loss on Metabolic Dysfunction Associated Steatohepatitis and Hepatic Fibrosis
Marina W. Takawy, Manal F. Abdelmalek
Current Diabetes Reports.2025;[Epub] CrossRef - Nanoparticle-based therapeutic strategies for chronic liver diseases: Advances and insights
Sathiyamoorthy Padmanaban, Ji-Won Baek, Sai Sahithya Chamarthy, Saipriya Chandrasekaran, Antony V Samrot, Vijayakumar Gosu, In-Kyu Park, Kamalakannan Radhakrishnan, Don-Kyu Kim
Liver Research.2025; 9(2): 104. CrossRef - Innovative Strategies in the Diagnosis and Treatment of Liver Cirrhosis and Associated Syndromes
Ashok Kumar Sah, Mohd Afzal, Rabab H. Elshaikh, Anass M. Abbas, Manar G. Shalabi, Pranav Kumar Prabhakar, Asaad M. A. Babker, Fariza Tursunbaevna Khalimova, Velilyaeva Aliya Sabrievna, Ranjay Kumar Choudhary
Life.2025; 15(5): 779. CrossRef - Poly(I:C)-Stimulated Exosomes Mitigate Cholesterol-Induced Hepatic Fibrosis by Modulating the TGF-β/Smad3 Signal Transduction Pathway
Shahla Asadizadeh, Parichehreh Yaghmaei, Nasim Hayati Roodbari, Azam Khedri
Hepatitis Monthly.2025;[Epub] CrossRef - The postbiotic ReFerm® versus standard nutritional support in advanced alcohol-related liver disease (GALA-POSTBIO): a randomized controlled phase 2 trial
Johanne K. Hansen, Mads Israelsen, Suguru Nishijima, Sara E. Stinson, Peter Andersen, Stine Johansen, Camilla D. Hansen, Maximilian Joseph Brol, Sabine Klein, Robert Schierwagen, Frank Erhard Uschner, Karolina Sulek, Ida F. Villesen, Katrine P. Lindvig, K
Nature Communications.2025;[Epub] CrossRef - ADVANCING CURCUMIN APPLICATIONS IN HEPATOCELLULAR CARCINOMA: INSIGHTS INTO PHARMACOKINETICS, PHARMACODYNAMICS AND NANODELIVERY SYSTEMS
RESHMA T. MATE, ANURADHA N. CHIVATE, NAMDEO R. JADHAV, NIRANJAN D. CHIVATE
International Journal of Applied Pharmaceutics.2025; : 19. CrossRef - Genetic Determinants of Apoptosis Regulate Liver Cirrhosis Risk: The FAS/FASL Polymorphism Connection in Iraqi Population
Wisam Hindawi Hoidy, Ahmed Ghdhban Al-Ziaydi
Indian Journal of Clinical Biochemistry.2025;[Epub] CrossRef - Histological and Molecular Evaluation of Liver Biopsies: A Practical and Updated Review
Joon Hyuk Choi
International Journal of Molecular Sciences.2025; 26(16): 7729. CrossRef - Resolving fibrosis by stimulating HSC-dependent extracellular matrix degradation
Sachin Sharma, Vijaya Prathigudupu, Carson Cable, Lia R. Serrano, Srilaxmi Nerella, Alina Chen, Ghmkin Hassan, Johnathon Lakins, Carlos Lizama Valenzuela, Tatsuya Tsukui, Roopa Ramamoorthi, Jae-Jun Kim, Holger Willenbring, Aras N. Mattis, Regan F. Volk, B
Science Translational Medicine.2025;[Epub] CrossRef - Association of ANGPT2 and NOS3 Gene Variants With Esophageal Variceal Progression in HCV‐Induced Cirrhosis
Omnia S. Nabih, Mohamed Abdel‐Samiee, Marwa A. Tahoon, Elaf Abozeid, Heba Demerdash, Fatma Omar Khalil, Randa M. Seddik, Marwa L. Helal, Gamalat A. El Gedawy, HalaA E. Khalil, Badawy W AboBakr. Yossef, Ahmed B. Zaid, Mai I. Elashmawy
Journal of Medical Virology.2025;[Epub] CrossRef - Сomponents of colostrum regulate the activity of the immune system in animals with liver fibrosis
E. G. Ivanov, E. M. Klimova, A. A. Bozhkov, M. K. Kovalova, A. I. Bozhkov
Regulatory Mechanisms in Biosystems.2025; 16(4): e25176. CrossRef - Emerging advanced approaches for diagnosis and inhibition of liver fibrogenesis
Tamer A. Addissouky, Majeed M. A. Ali, Ibrahim El Tantawy El Sayed, Yuliang Wang
The Egyptian Journal of Internal Medicine.2024;[Epub] CrossRef - Inhibition of hepatic stellate cell activation by nutraceuticals: an emphasis on mechanisms of action
Vasudevan Sekar, Venkateish VP, Vani Vijay, Annapoorna BR, Nivya Vijayan, Madan Kumar Perumal
Journal of Food Science and Technology.2024; 61(11): 2046. CrossRef - The Role of Macrophage Inhibitory Factor in TAA-Induced Liver Fibrosis in Mice: Modulatory Effects of Betaine
Tatjana Radosavljevic, Dusan Vukicevic, Jasmina Djuretić, Kristina Gopcevic, Milica Labudovic Borovic, Sanja Stankovic, Janko Samardzic, Milica Radosavljevic, Danijela Vucevic, Vladimir Jakovljevic
Biomedicines.2024; 12(6): 1337. CrossRef - Fibrosis and Hepatocarcinogenesis: Role of Gene-Environment Interactions in Liver Disease Progression
Anindita Banerjee, Patrizia Farci
International Journal of Molecular Sciences.2024; 25(16): 8641. CrossRef - Multiomic predictors for regression of cirrhosis: Clinical implications and future directions
Binghua Li, Decai Yu
iLIVER.2024; 3(4): 100116. CrossRef - Commonly encountered symptoms and their management in patients with cirrhosis
Cyriac Abby Philips
Frontiers in Medicine.2024;[Epub] CrossRef - Antioxidant Potential of Xanthohumol in Disease Prevention: Evidence from Human and Animal Studies
Jakub Piekara, Dorota Piasecka-Kwiatkowska
Antioxidants.2024; 13(12): 1559. CrossRef - AdhMMP8 Vector Administration in Muscle: An Alternate Strategy to Regress Hepatic Fibrosis
Jesús García-Bañuelos, Edén Oceguera-Contreras, Ana Sandoval-Rodríguez, Blanca Estela Bastidas-Ramírez, Silvia Lucano-Landeros, Daniela Gordillo-Bastidas, Belinda C. Gómez-Meda, Arturo Santos, Eira Cerda-Reyes, Juan Armendariz-Borunda
Cells.2023; 12(17): 2127. CrossRef - Nutritional deficiency in patients with liver cirrhosis
Maria S. Zhigalova, Vladimir V. Kiselev, Alla A. Ryk, Petr A. Yartsev
Clinical nutrition and metabolism.2023; 4(4): 265. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
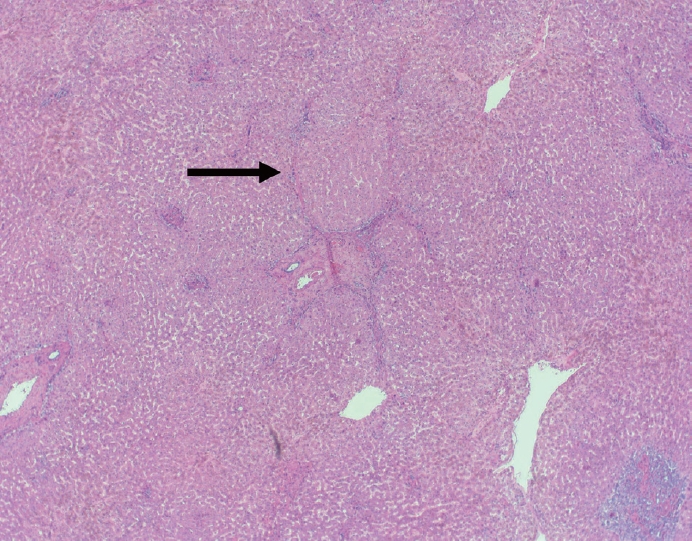
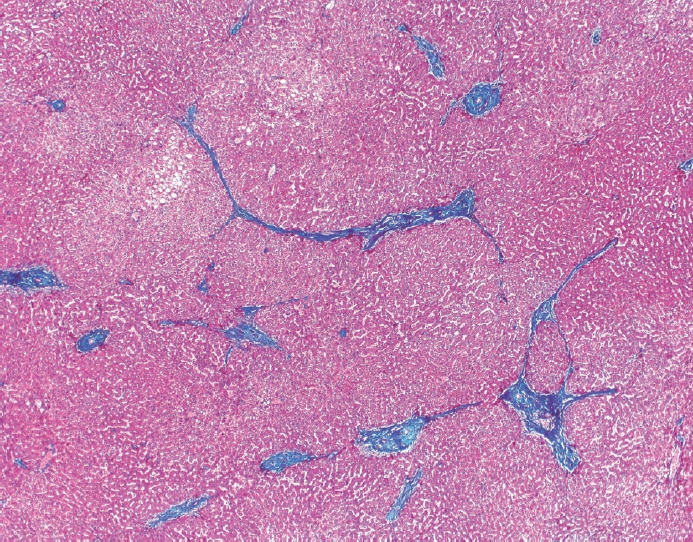
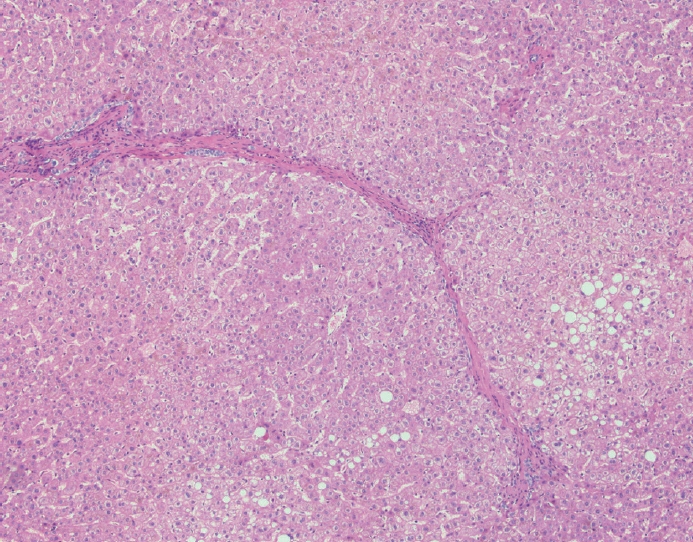
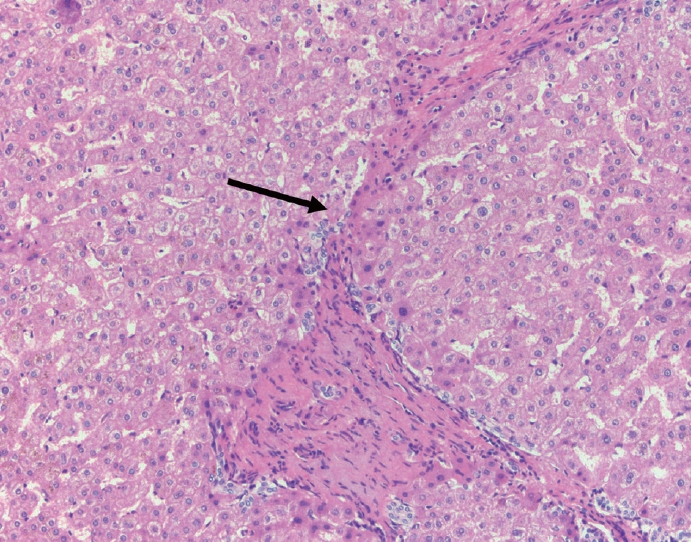
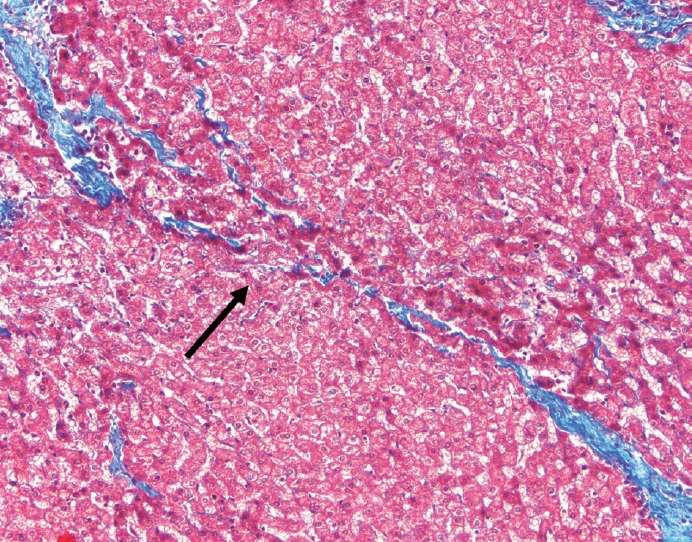
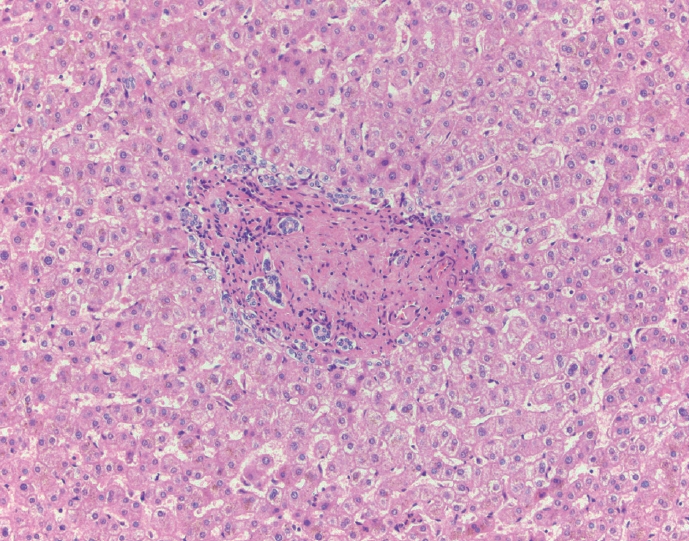
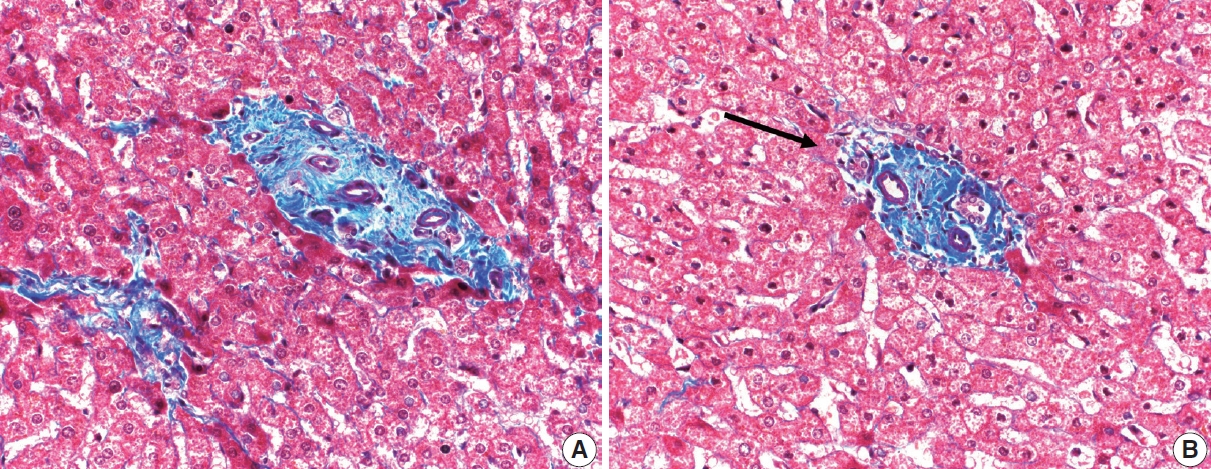







Fig. 1.
Fig. .
Fig. 3.
Fig. 4.
Fig. 5.
Fig. 6.
Fig. 7.
| Fibrosis | Score |
|---|---|
| None | 0 |
| Portal fibrosis confined to the portal tract | 1 |
| Periportal fibrosis or portal to portal fibrosis with intact architecture | 2 |
| Bridging fibrosis with architectural distortion but no cirrhosis | 3 |
| Probably or definitive cirrhosis | 4 |
| Description | ||
|---|---|---|
| Hepatitis assessment | ||
| Inactive | Portal inflammation only or rare foci of interface or lobular hepatitis, no confluent necrosis | |
| Active, non-severe | Varying degrees of interface and lobular hepatitis easily identified at low power, no confluent necrosis | |
| Active, severe | Confluent necrosis (perivenular drop out or bridging necrosis or parenchymal collapse) | |
| Fibrosis stage | ||
| Early | No fibrosis or portal fibrosis | |
| Intermediate | Fibrous septa, focal or frequent | |
| Advanced | Fibrous septa with focal or diffuse nodularity (developing or established cirrhosis) | |
| P-I-R fibrosis quality | ||
| Predominantly progressive | Most of the specimen shows progressive forms of stroma | |
| Indeterminate | Uncertain mix or balance between progressive and regressive forms of stroma | |
| Predominantly regressive | Most of the specimen shows regressive forms of stroma | |

