 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 49(6); 2015 > Article
-
Review
The Continuing Value of Ultrastructural Observation in Central Nervous System Neoplasms in Children - Na Rae Kim, Sung-Hye Park1
-
Journal of Pathology and Translational Medicine 2015;49(6):427-437.
DOI: https://doi.org/10.4132/jptm.2015.09.19
Published online: October 13, 2015
Department of Pathology, Gachon University Gil Medical Center, Incheon, Korea
1Department of Pathology, Seoul National University College of Medicine, Seoul, Korea
- Corresponding Author: Na Rae Kim, MD Department of Pathology, Gachon University Gil Medical Center, 21 Namdong-daero 774 beon-gil, Namdong-gu, Incheon 21565, Korea Tel: +82-32-460-3847 Fax: +82-32-460-2394 E-mail:clara_nrk@gilhospital.com
• Received: September 15, 2015 • Accepted: September 19, 2015
© 2015 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 10,980 Views
- 78 Download
- 1 Scopus
- Abstract
- SPINDLE CELL TUMORS OF THE CENTRAL NERVOUS SYSTEM
- SMALL ROUND CELL CATEGORY OF PEDIATRIC CENTRAL NERVOUS SYSTEM TUMORS
- OLIGODENDROGLIOMA AND OLIGODENDROGLIOMA-LIKE TUMORS
- PAPILLARY TUMORS OF THE CENTRAL NERVOUS SYSTEM
- RHABDOID CELL TUMORS
- PLEOMORPHIC CELL TUMORS
- CONCLUSION
- NOTES
- Acknowledgments
- REFERENCES
Abstract
- Central nervous system (CNS) neoplasms are the second most common childhood malignancy after leukemia and the most common solid organ neoplasm in children. Diagnostic dilemmas with small specimens from CNS neoplasms are often the result of multifactorial etiologies such as frozen or fixation artifact, biopsy size, or lack of knowledge about rare or unfamiliar entities. Since the late 1950s, ultrastructural examination has been used in the diagnosis of CNS neoplasms, though it has largely been replaced by immunohistochemical and molecular cytogenetic studies. Nowadays, pathologic diagnosis of CNS neoplasms is achieved through intraoperative cytology, light microscopy, immunohistochemistry, and molecular cytogenetic results. However, the utility of electron microscopy (EM) in the final diagnosis of CNS neoplasms and investigation of its pathogenetic origin remains critical. Here, we reviewed the distinguishing ultrastructural features of pediatric CNS neoplasms and emphasize the continuing value of EM in the diagnosis of CNS neoplasms.
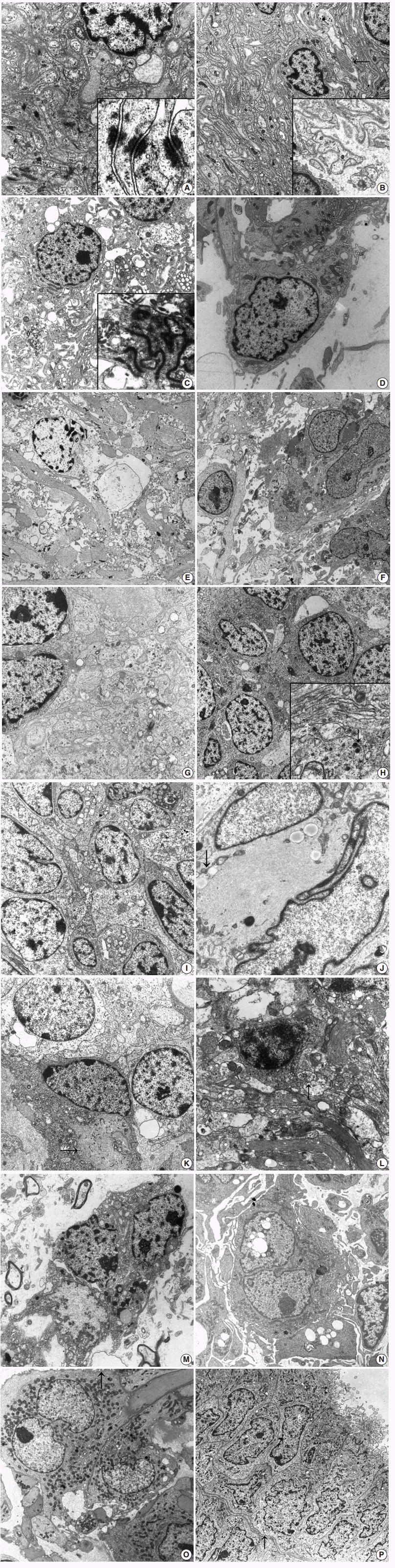
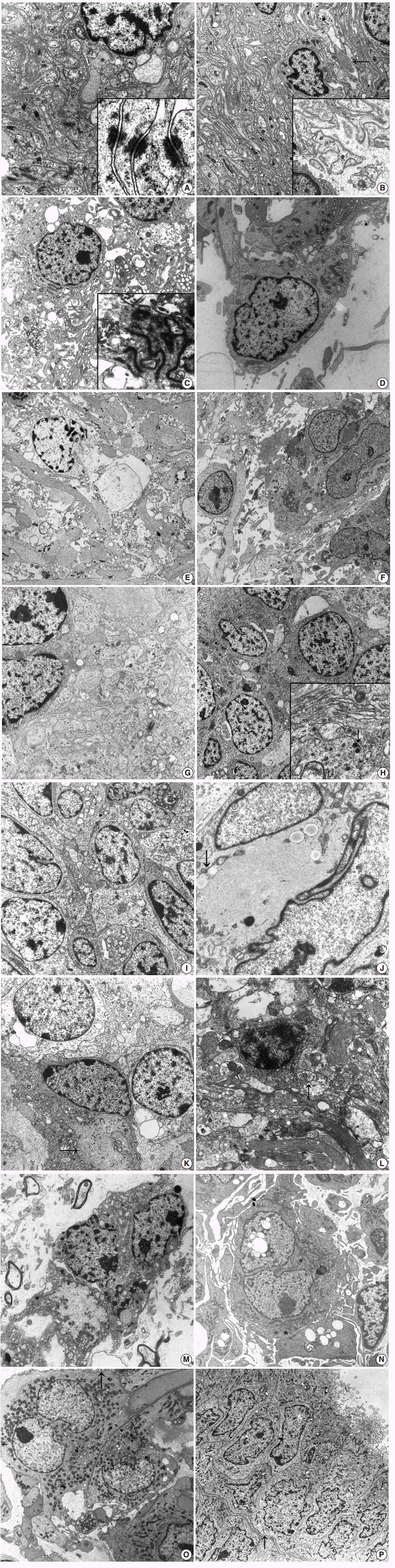
- First, the spindle cell category includes the following entities: meningioma, schwannoma, ependymoma, and astrocytoma including pilocytic astrocytoma, and pilomyxoid astrocytoma. Among these, ependymoma and pilocytic astrocytoma are common in children, while meningioma or schwannoma are not common. Rather, meningioma associated with meningioangiomatosis has been reported mainly in childhood [5]. Previous reports on meningioma arising in meningioangiomatosis describe entrapped neurons in the infiltrating meningioma [15]. Ultrastructural findings of meningioma show its meningothelial arachnoidal cell nature, i.e., epithelial and mesenchymal nature; connective tissue fibers and basal lamina are not usually seen among tumor cells within the syncytium (Fig. 1A). Elongated interdigitating cell processes filled with copious amounts of intracytoplasmic intermediate filaments show an interdigitating and jigsaw pattern, corresponding to the whorls of enwrapping meningioma tumor cells under light microscopy. The epithelial nature, such as well-formed desmosomes, is a characteristic ultrastructural feature. The extracellular space of meningioma sometimes contains fine granular materials, mimicking basal lamina. Meningiomas also show basement membrane separating the syncytium from the fibrous septum or the perivascular connective tissue [16]. Rhabdoid or chordoid meningioma can have a thick continuous basal lamina [17-19]. Schwannoma, also called neurilemmoma, neuroma, or neurinoma, is relatively rare in the brain with or without cranial nerve-relation. Intracerebral schwannomas not related to cranial nerves are rare, and most cases occur in the first two decades of life [20]. Schwannoma can be diagnosed in children who might be associated with neurofibromatosis type 2, while intracerebral schwannomas have a weak relationship with neurofibromatosis type 2. The ultrastructure of schwannoma is almost exclusively cells with characteristics of differentiated Schwann cells of a neuroectodermal origin. Numerous finger-like cytoplasmic processes are lined by continuous basal lamina with occasional duplication (Fig. 1B). Closely apposed stacks of plasma membrane, i.e., reminiscent of myelin lamellae, are characteristic of schwannoma. Compared to normal collagen fibers of 64-nm periodicity, so-called Luse bodies show more widely spaced collagen fibers with up to 150-nm periodicity and are frequently found in schwannoma. The tumor cells themselves are composed of cigar-shaped or elliptical nuclei containing one or two small nucleoli and rare nuclear bodies. The cytoplasm contains well-developed Golgi complexes, scattered mitochondria, short segments of rough endoplasmic reticulums (RERs), small numbers of ribosomes, and scattered polysomes. Absence of pinocytotic vesicles always happens in schwannoma. Microfilaments, microtubules, and small vesicles are often found, but few mitochondria and ribosomes are found. These ultrastructural characteristics of schwannoma provide diagnostic clues, particularly regarding small cell changes in schwannoma or small cell malignant peripheral nerve sheath tumors [21,22]. Ependymoma occurs mainly in the ventricular system, and infratentorial ependymomas occur predominantly in children, although development of ependymomas can occur at any age [23-25]. Ultrastructural findings of a well-developed ependymal tumor, which is likely derived from ependymal cells lining the CNS ventricular system, show both epithelial and astrocytic features, and the tumor cells of grade I and II ependymal tumors resemble typical ependymocytes, while anaplastic ependymomas are poorly differentiated with minimal evidence of such ependymal differentiation. Ependymal differentiation includes well-developed junctions, such as zonula adherens or occludens, villous projections, and cilia in a 9+2 arrangement of microtubules (Fig. 1C). The centriole or blepharoplast is located in the basis of the cilia; in the cytoplasm, microtubules or glial filaments are present in the perikaryal area or cytoplasmic processes. Basal lamina is observed in the ependymoma and around blood vessels but not around tumor cells [26]. However, all of the above-mentioned findings are not specific for ependymomas; these ultrastructural findings are shared by angiocentric glioma, i.e., monomorphous angiocentric neuroepithelial tumor, composed of bipolar spindle cells and epithelioid round cells although the occurrence of angiocentric glioma has not yet been reported in children [9]. Angiocentric glioma is composed of infiltrating round, monopolar epithelioid cells arranged in a perivascular arrangement and bipolar spindle-shaped glial cells. Epithelioid monopolar cells showing microlumina filled with numerous microvilli and some cilia with a 9+2 or abnormal 10+2 configuration. Intermediate junctions of complex interdigitating membranes and basal lamina have also been observed [27]. Ependymomas and angiocentric glioma share molecular cytogenetic alterations, as indicated by shared ultrastructural features [28]. These findings suggest that angiocentric glioma belongs to a lineage of ependymal tumors.
- Pilocytic astrocytoma is a commonly encountered CNS tumor in children, and the most common site of occurrence is the sellar region. In contrast, pituitary adenoma, meningioma, and metastatic carcinoma are common tumors in adults but are rarely encountered in children.1 Pilomyxoid astrocytoma and pilocytic astrocytoma share pathogenetic and clinicopathologic features; BRAF oncogene activation through KIAA1549-BRAF fusion most commonly found in pilocytic astrocytoma has also been described in pilomyxoid astrocytoma [29,30]. As with light microscopy, irregularly shaped, amorphous, granular electrondense Rosenthal fibers are commonly found in pilocytic astrocytoma under EM [31]. These Rosenthal fibers are closely located in the glial intermediate filaments [32]. Pilomyxoid astrocytoma showing a monomorphic histologic appearance more commonly occurs in infants and young children compared with pilocytic astrocytoma [33]. Ultrastructurally, pilomyxoid astrocytomas have two morphologically distinct cell types [34]; one is spindle cells showing neurite-like features, and the other is spindle cells showing overt astrocytic morphology (Fig. 1D). Despite shared histologic and molecular cytogenetic findings, pilomyxoid astrocytoma relating to pilocytic astrocytoma and other glioneuronal tumors remain undefined and controversial. With progression of WHO grade in astrocytic tumors, there are sparser glial filaments and more irregular microvillus-like cell projections in anaplastic astrocytoma or glioblastoma, compared to diffuse astrocytoma (Fig. 1E, F). Pediatric glioblastomas are rarely encountered [35-37]. Excluding components of gliosarcoma, pediatric brain sarcomas are rare. However, various types of primary intracranial sarcomas can occur in the absence of radiation history [38,39]. Although rare cases of rhabdomyosarcoma or chondrosarcoma have also been described, most reported cases in children are fibrosarcomas [40]. Proliferation of spindle cells resembling meningioma on immunohistochemistry and light microscopy but with a classical herring-bone pattern suggests fibrosarcoma. Fibrosarcoma is composed of fibroblast-like spindle cells with well-developed, abundant dilated RERs and prominent Golgi complexes [41]. Small patches of basal lamina materials lining some tumor cell surfaces are observed in rare cases. Features of myoid differentiation have not been observed.
SPINDLE CELL TUMORS OF THE CENTRAL NERVOUS SYSTEM
- In this group, small round cell pediatric CNS tumors include medulloblastoma, CNS primitive neuroectodermal tumor (PNET)/embryonal tumor with abundant neuropils and true rosettes, atypical teratoid and rhabdoid tumor (AT/RT), and neuroblastoma [2,42-48]. Medulloblastoma is a common malignant primary brain tumor in children, categorized as a PNET of the cerebellum, i.e., an undifferentiated tumor exhibiting classic undifferentiated multipotent tumor cells showing myoblastic, neuronal, glial, and melanotic differentiation observed under light microscopy, immunohistochemistry, and EM [48]. A broad spectrum of neural differentiation has been identified in nearly all cases of medulloblastoma [49]. Therefore, there is a spectrum in which the most undifferentiated area might have large undifferentiated cells compared to more differentiated areas exhibiting neuroblastic differentiation. Ultrastructurally, medulloblastoma is composed of primitive cells having a high nuclear-cytoplasmic ratio, and those cells that lack distinguishing ultrastructural findings such as tightly packed poorly differentiated oval to polygonal tumor cells with large nuclei; Rosenthal fibers, i.e., astrocytic differentiation; or neuronal differentiation such as rudimentary synaptic junctions or dense core granules (Fig. 1G). The central acellular area is composed of centrally projected broad cell processes, forming Homer-Wright rosettes. These central acellular rosettes composed of cytoplasmic processes are found in other tumors such as neuroblastoma, neurocytoma, and ependymoma [25,50,51]. Glial intermediate filaments fill the cell processes in an ependymoma or medulloblastoma with glial differentiation, while microtubules are found in tumors of neuronal differentiation such as neurocytoma or neuroblastoma (Fig. 1H) [49,51,52].
- PNET was originally described as an undifferentiated neuroepithelial tumor with mesenchymal differentiation and now encompasses tumors showing pluripotentiality toward neuronal, astrocytic, and other cell lines such as those of a mesenchymal lineage (Fig. 1I). However, this entity might be a confusing and controversial factor in the diagnosis and classification of pediatric embryonal tumors. Some cases of PNET mimicking AT/RT have been reported [53]. AT/RT, which occurs mainly in children under the age of 3 years and is associated with frequent recurrence, is a malignant rhabdoid tumor (MRT), i.e., a CNS counterpart of renal MRT, belonging to rhabdoid tumor predisposition syndrome. Mutation or deletion of SMARCB1/INI-1/hSNF5/BAF47-tumor-suppressor gene located on chromosome band 22q11.2 inactivates the tumor suppressor gene, resulting in subsequent negative immunostaining in AT/RT but not in other rhabdoid phenotypes of CNS neoplasms, although mutations of INI-1 in some choroid plexus carcinomas have been reported [47,53,54]. Immunohistochemistry for INI-1 is actually superior to EM or small biopsy of light microscopy because ultrastructural descriptions of AT/RT are few and lack specific CNS cell components of glial or neuronal lineage (Fig. 1J). Histologically, AT/RT shows various mixed histologies of epithelial, mesenchymal, rhabdoid, and primitive neuroepithelial components, suggesting an origin of immature and pluripotent neuroectodermal cells capable of differentiating along multiple lineages, as demonstrated by EM [55].
SMALL ROUND CELL CATEGORY OF PEDIATRIC CENTRAL NERVOUS SYSTEM TUMORS
- Oligodendroglioma-like tumors, i.e., oligodendroglioma-mimickers, include oligodendroglioma, dysembryoplastic neuroepithelial tumor (DNT), neurocytoma, clear cell ependymoma (CCE), ganglioglioma, and gangliocytoma [25,56-58]. Oligodendroglioma is composed of closely apposed round-shaped tumor cells with euchromatic round nuclei and cytoplasm containing some microtubules and few glial filaments (Fig. 1K). Short broad cell processes might be seen within the tumor cells. Cell junctions or surface microvilli are not found in oligodendroglioma [25,59]. Among oligodendroglioma-mimickers, neurocytomas have distinct immunohistochemical and ultrastructural characteristics compared with oligodendrogliomas [50,51,60]. Both can have microtubules, but the former exhibit neuronal differentiation such as synaptic vesicles or dense core granules. Central and extraventricular neurocytoma has been reported as ependymoma of the foramen of Monro or intraventricular oligodendroglioma due to mimicry of oligodendroglioma-like cells under light microscopy [60]. Characteristic immunohistochemical findings of rosette-like acellular fibrillary areas stained with neuronal markers such as synaptophysin or NeuN might not be identified in extraventricular neurocytomas [61]. Ultrastructurally, neurocytoma has numerous thin neuritic processes filled with dense core granules and microtubules with or without synapses (Fig. 1L). DNT consists of neoplastic oligodendroglial-like cells (OLCs) and elongated processes forming a neuropil-like structure in the mucinous extracellular area [58]. Ultrastructurally, OLCs have round to oval or elongated nuclei and scanty cytoplasm filled with electron-lucent synaptic vesicles, whereas elongated tumor cells have abundant cellular processes containing scanty microtubules and dense-core granules (Fig. 1M). OLCs of DNT can be distinguished from astrocytes by the lack of glial intermediate filaments and from neuronal cells by the presence of microtubules. Some cells of DNT have cytoplasmic ribosome-lamellae complexes, also found in lymphoid neoplasms or glioblastomas [62,63]. These structures have been regarded as astrocytic differentiation but remain elusive. CCE resembling oligodendroglioma and neurocytic tumor under light microscopy can be distinguished from the latter using EM, which shows intracytoplasmic lumen lined by microvilli, complex cell junctions, and cilia. The clear cell morphology of CCE is caused by organellefree areas of the cytoplasm with edema and vacuolization [64,65].
- Ganglioglioma and gangliocytoma are composed of several types of tumor cells; neuronal tumor cells have dense core granules, diagnostically salient features, and synaptic junctions. Spherical protein bodies are found in gangliogliomas [56]. Multivesicular bodies, abundant autophagic vacuoles, and dense core vesicles are seen in neoplastic neuronal cells of ganglioglioma and gangliocytomas [66].
OLIGODENDROGLIOMA AND OLIGODENDROGLIOMA-LIKE TUMORS
- Papillary tumors of the CNS observable under light microscopy include PGNT, tumors of the suprasellar area such PTPR, pituitary adenoma of papillary configuration, or chordoid glioma of the third ventricle [67-69]. PGNTs can occur at any age, and pediatric cases have been reported [67]. Ultrastructurally, either astrocytic or neuronal differentiation and poorly differentiated or uncommitted primitive cells are found in PGNT [52,69]. PTPR was recently added to the 2007 WHO classification as a rare neuroepithelial tumor of papillary appearance arising in the pineal area [45,46]. This tumor has been named papillary pineocytoma or choroid plexus papilloma due to papillary features and epithelial morphology showing immunopositivity for pancytokeratin and ependymal differentiation. Pediatric occurrence of PTPR is rare. Specialized ependymal cells of the subcommissural organ (SCO) are considered the origin of PTPR because of ultrastructural findings of divergent differentiation of epithelial, ependymal, and neuroendocrine cells. On EM, clear and dark epithelioid cells have an ovoid nuclei and abundant cytoplasm filled with abundant organelles including RERs, dense core vesicles, microtubules, numerous clear and coated vesicles, mitochondria, and intermediate filaments [70]. Abundant microvilli and less frequent cilia are found at the apical pole (Fig. 1N). Well-formed junctional complexes are found at the apical portion, and the basal portion is surrounded by a basement membrane. Chordoid glioma of the third ventricle is characterized by clusters and cords of epithelioid cells in a mucinous chordoid matrix. It is a slowly growing glial tumor occurring mainly in adults, although rare cases of pediatric choroid glioma have been reported [68,71]. Chordoid glioma and PTPR share an anatomic location, i.e., circumventricular organ occurrence and subsequent shared ultrastructural findings; secretory vesicles as well as cilia, microvilli, and junctional complexes such as hemidesmosomes, are suggestive of the origin of modified specialized ependymal, i.e., tanycytes of the SCO such as lamina terminalis (Fig. 1O) [68,69]. Choroid plexus papilloma shows cuboidal-shaped tumor cells with apical-basal orientation with varying sizes of apical club-like or roundish microvilli filled with glycogen particles and rare cilia with a 9+2 arrangement of microtubules (Fig. 1P) [25,72,73]. The neoplastic cells contain numerous free ribosomes, glycogen granules, and RERs. Elongated junctional complexes were occasionally seen near the apical ends. The basal portions of the tumor cells are lined with a continuous basal lamina. These apical cilia or glycogen particles are more often seen in infantile choroid plexus papillomas than in those of older age [72].
PAPILLARY TUMORS OF THE CENTRAL NERVOUS SYSTEM
- Rhabdoid cells are designated as large cells with eccentrically located nuclei and well-demarcated abundant, eosinophilic globular inclusions with characteristic immunoreactivity for vimentin, keratin, and epithelial membrane antigen, which ultrastructurally correspond to paranuclear whorls of intermediate filaments. These rhabdoid changes can appear in various neoplasms from diverse organ sites [54,73-75]. In the CNS, most rhabdoid tumors occur in the infratentorial posterior fossa of very young children. Histological diagnosis of an MRT depends on identification of characteristic rhabdoid choroid plexus carcinoma, an extremely rare variant of choroid plexus carcinoma, belonging to choroid plexus tumor; however, carcinoma can lose its papillary pattern, and this ill-defined growth pattern with small foci of epithelial differentiation can make diagnosis challenging and mimic AT/RT due to overlapping morphological, immunohistochemical, and ultrastructural features, except for INI-1 expression [54,55,74]. Like other MRT, EM shows the typical rhabdoid cells with paranuclear whorls or bundles of intermediate filaments as well as reminiscent retention of choroid plexus differentiation [73]. These rhabdoid features can be seen in rhabdoid meningioma, ependymoma, glioma, or germ cell tumors and even in metastatic rhabdoid melanoma [19,75,76].
RHABDOID CELL TUMORS
- CNS tumors showing pleomorphic features under light microscopy include pleomorphic xanthoastrocytoma (PXA), glioblastoma, AT/RT, or undifferentiated primitive tumors [63,77,78]. Tumors such as AT/RT or undifferentiated primitive tumors were previously discussed in the section on small round cell tumors. PXA that is prevalent in children belongs to an astrocytic lineage and appears as irregularly-shaped heterochromatic nuclei and abundant cytoplasm filled with lysosomes, lipid droplets, and glial filaments [63]. Ultrastructurally, PXA is composed of two types of cells; spindle-shaped astrocytic cells and bizarre giant cells filled with glial filaments and lipid droplets; the astrocytic tumor undergoes neuronal differentiation. Basal lamina surrounds the individual tumor cells or in groups, in contrast to other astrocytic tumors. Cytoplasmic lysosomes and ribosomelamellae complexes are predominant in PXA, as in DNT, lymphoid leukemia and glioblastoma [62,63].
PLEOMORPHIC CELL TUMORS
- Despite declining usage, EM retains its diagnostic usefulness in neuropathology, particularly in distinguishing histological look-alikes, poorly differentiated or undifferentiated CNS neoplasms. When there is an overlap in the immunohistochemical profiles between CNS tumors, ultrastructural examination serves to confirm the concrete diagnosis even if the components of surrounding non-neoplastic cells may be misinterpreted as those of the tumor cells.
- In this article, we summarized and reviewed diagnostic ultrastructural findings of CNS neoplasms commonly found in children.
CONCLUSION
Acknowledgments
Fig. 1. (A) Meningioma shows closely apposed oval- to spindle-shaped tumor cells having interdigitating cell processes lined by well-formed desmosomes (×8,000). Inset indicates tonofibrils attached to the desmosomal plaques (×30,000). (B) Schwannoma shows abundant reduplicated continuous basal lamina surrounding the interdigitating cell processes (×3,500). Note the extracellular long-spacing collagen (arrow) and high power view of reduplicated basal lamina (inset, ×6,500). (C) Ependymoma reveals spindle cells with numerous cytoplasmic processes filled with bundles of intermediate filaments (×4,000). Inset shows well-formed zonula occludens with surface microvilli (×15,000). (D) Pilomyxoid astrocytoma shows bipolar cells with surface microvilli and cilia in the electron-lucent extracellular space (×9,000). Note some intracellular microlumen. (E) Astrocytoma shows loosely scattered round to oval cells having numerous long cytoplasmic processes filled with bundles of glial filaments and scarce other organelles (×2,500). (F) Glioblastoma shows many pleomorphic spindle and oval cells with numerous cytoplasmic processes containing bundles of intermediate filaments with surface microvilli-like differentiation (×2,500). (G) Medulloblastoma shows that loosely arranged oval-shaped tumor cells project cytoplasmic processes forming rosettes filled with glial filaments and dense core granules indicating neuroglial differentiation (×9,000). (H) Neuroblastoma reveals that closely packed polygonal to spherical tumor cells have numerous, thin, electron-lucent cell processes forming an interlacing meshwork between groups of cell bodies (×9,000). Inset shows longitudinally-oriented 20 nm microtubules (white arrow) and dense core granules (black arrow, ×15,000). (I) Primitive neuroectodermal tumor reveals round to closely apposed oval-shaped tumor cells with abundant cytoplasmic processes containing sparse organelles including glycogen particles, mitochondria, and some microtubules (×8,000).(J) Atypical teratoid and rhabdoid tumor reveals paranuclear aggregates of intermediate filaments (arrow) compressing the heterochromatic nuclei (×15,000). (K) Oligodendroglioma presents tumor cells with uniform round nuclei and sparse cytoplasmic organelles as well as some irregularly shaped cell processes (arrow) containing occasional globoid collections of intermediate filaments (×3,500). (L) Neurocytoma reveals round tumor cells with a moderate amount of cytoplasm and numerous long thin cell processes containing microtubules, few electron dense core granules and secretory vesicles (arrow), and glial intermediate filaments (×8,000). (M) Dysembryoplastic neuroepithelial tumor reveals oligodendroglial-like cells with elongated bulbous cell processes forming a neuropil-like structure filled with intermediate filaments in electron-lucent mucoid extracellular spaces (×7,000).(N) Papillary tumor of the pineal region reveals polygonal-shaped tumor cells having short-villous cell surfaces and cytoplasm having abundant rough endoplasmic reticulums with distended cisternae and vacuoles (×3,500). (O) Chordoid glioma of the third ventricle reveals tumor cells with cytoplasmic processes filled with intermediate glial filaments and surrounded by basal lamina (×7,000). Note the numerous surface microvilli (arrow). (P) Choroid plexus papilloma shows ovoid to polygonal tumor cells arranged in glandular patterns joined by well-formed junctional complexes and a continuous basal lamina (arrow) surrounding these glandular structures (×1,500). Note the apical portion of tumor cells lined by numerous microvilli and cilia composed of 9+2 microtubules.
- 1. Papathoma P, Thomopoulos TP, Karalexi MA, et al. Childhood central nervous system tumours: incidence and time trends in 13 Southern and Eastern European cancer registries. Eur J Cancer 2015; 51: 1444-55. ArticlePubMed
- 2. Schmidt LS, Schmiegelow K, Lahteenmaki P, et al. Incidence of childhood central nervous system tumors in the Nordic countries. Pediatr Blood Cancer 2011; 56: 65-9. ArticlePubMedPDF
- 3. Gerstner ER, Batchelor TT. Primary central nervous system lymphoma. Arch Neurol 2010; 67: 291-7. ArticlePubMed
- 4. Zhang C, Bao Z, Zhang W, Jiang T. Progress on molecular biomarkers and classification of malignant gliomas. Front Med 2013; 7: 150-6. ArticlePubMedPDF
- 5. Kim NR, Cho SJ, Suh YL. Allelic loss on chromosomes 1p32, 9p21, 13q14, 16q22, 17p, and 22q12 in meningiomas associated with meningioangiomatosis and pure meningioangiomatosis. J Neurooncol 2009; 94: 425-30. ArticlePubMedPDF
- 6. Ahsan J, Hashmi SN, Muhammad I, et al. Spectrum of central nervous system tumours: a single center histopathological review of 761 cases over 5 years. J Ayub Med Coll Abbottabad 2015; 27: 81-4. PubMed
- 7. Pastuszak Z˙, Tomczykiewicz K, Piusin’ska-Macoch R, Ste¸pien’ A, Kordowska J. The occurrence of tumors of the central nervous system in a clinical observation. Pol Merkur Lekarski 2015; 38: 88-92. PubMed
- 8. Hicks J, Mierau GW. The spectrum of pediatric tumors in infancy, childhood, and adolescence: a comprehensive review with emphasis on special techniques in diagnosis. Ultrastruct Pathol 2005; 29: 175-202. ArticlePubMed
- 9. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO classification of tumours of the central nervous system. 4th ed. Lyon: IARC Press, 2007.
- 10. Langford LA. Central nervous system neoplasms: indications for electron microscopy. Ultrastruct Pathol 1996; 20: 35-46. ArticlePubMed
- 11. Uematsu Y. The role of electron microscopy in the diagnosis of surgical pathology in the central nervous system. Med Mol Morphol 2006; 39: 127-35. ArticlePubMedPDF
- 12. Kleihues P, Cavenee WK. World Health Organization classification of tumours of pathology and genetics of tumours of the nervous system. 3rd ed. Lyon: IARC Press, 2000.
- 13. Speirs CK, Simpson JR, Robinson CG, et al. Impact of 1p/19q codeletion and histology on outcomes of anaplastic gliomas treated with radiation therapy and temozolomide. Int J Radiat Oncol Biol Phys 2015; 91: 268-76. ArticlePubMed
- 14. Mur P, Mollejo M, Hernández-Iglesias T, et al. Molecular classification defines 4 prognostically distinct glioma groups irrespective of diagnosis and grade. J Neuropathol Exp Neurol 2015; 74: 241-9. ArticlePubMed
- 15. Giangaspero F, Guiducci A, Lenz FA, Mastronardi L, Burger PC. Meningioma with meningioangiomatosis: a condition mimicking invasive meningiomas in children and young adults: report of two cases and review of the literature. Am J Surg Pathol 1999; 23: 872-5. PubMed
- 16. Kepes J. Electron microscopic studies of meningiomas. Am J Pathol 1961; 39: 499-510. PubMedPMC
- 17. Donato G, Ferraro G, Signorelli F, et al. Chordoid meningioma: case report and literature review. Ultrastruct Pathol 2006; 30: 309-14. ArticlePubMed
- 18. Couce ME, Aker FV, Scheithauer BW. Chordoid meningioma: a clinicopathologic study of 42 cases. Am J Surg Pathol 2000; 24: 899-905. PubMed
- 19. Buccoliero AM, Castiglione F, Rossi Degl’Innocenti D, et al. Pediatric rhabdoid meningioma: a morphological, immunohistochemical, ultrastructural and molecular case study. Neuropathology 2011; 31: 59-65. ArticlePubMed
- 20. Srinivas R, Krupashankar D, Shasi V. Intracerebral schwannoma in a 16-year-old girl: a case report and review of the literature. Case Rep Neurol Med 2013; 2013: 171494.ArticlePubMedPMCPDF
- 21. Peydró-Olaya A, Llombart-Bosch A, Carda-Batalla C, Lopez-Guerrero JA. Electron microscopy and other ancillary techniques in the diagnosis of small round cell tumors. Semin Diagn Pathol 2003; 20: 25-45. PubMed
- 22. Kim NR, Ha SY, Cho HY. Utility of transmission electron microscopy in small round cell tumors. J Pathol Transl Med 2015; 49: 93-101. ArticlePubMedPMCPDF
- 23. Mrak RE. The Big Eye in the 21st century: the role of electron microscopy in modern diagnostic neuropathology. J Neuropathol Exp Neurol 2002; 61: 1027-39. ArticlePubMed
- 24. Langford LA, Barré GM. Tanycytic ependymoma. Ultrastruct Pathol 1997; 21: 135-42. ArticlePubMed
- 25. Scheithauer BW, Bruner JM. Central nervous system tumors. Clin Lab Med 1987; 7: 157-79. ArticlePubMed
- 26. Baloyannis SJ, Baloyannis IS. The fine structure of ependymomas. CNS Oncol 2014; 3: 49-59. ArticlePubMedPMC
- 27. Miyata H, Ryufuku M, Kubota Y, Ochiai T, Niimura K, Hori T. Adult-onset angiocentric glioma of epithelioid cell-predominant type of the mesial temporal lobe suggestive of a rare but distinct clinicopathological subset within a spectrum of angiocentric cortical ependymal tumors. Neuropathology 2012; 32: 479-91. ArticlePubMed
- 28. Preusser M, Hoischen A, Novak K, et al. Angiocentric glioma: report of clinico-pathologic and genetic findings in 8 cases. Am J Surg Pathol 2007; 31: 1709-18. PubMed
- 29. de Chadarevian JP, Halligan GE, Reddy G, et al. Glioneuronal phenotype in a diencephalic pilomyxoid astrocytoma. Pediatr Dev Pathol 2006; 9: 480-7. ArticlePubMedPDF
- 30. Horbinski C, Nikiforova MN, Hagenkord JM, Hamilton RL, Pollack IF. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol 2012; 14: 777-89. ArticlePubMedPMC
- 31. Liberski PP. The ultrastructure of glial tumors of astrocytic lineage: a review. Folia Neuropathol 1998; 36: 161-77. PubMed
- 32. Wippold FJ 2nd, Perry A, Lennerz J. Neuropathology for the neuroradiologist: Rosenthal fibers. AJNR Am J Neuroradiol 2006; 27: 958-61. PubMedPMC
- 33. Paraskevopoulos D, Patsalas I, Karkavelas G, Foroglou N, Magras I, Selviaridis P. Pilomyxoid astrocytoma of the cervical spinal cord in a child with rapid progression into glioblastoma: case report and literature review. Childs Nerv Syst 2011; 27: 313-21. ArticlePubMedPDF
- 34. Fuller CE, Frankel B, Smith M, et al. Suprasellar monomorphous pilomyxoid neoplasm: an ultastructural analysis. Clin Neuropathol 2001; 20: 256-62. PubMed
- 35. Kim NR, Wang KC, Bang JS, et al. Glioblastomatous transformation of ganglioglioma: case report with reference to molecular genetic and flow cytometric analysis. Pathol Int 2003; 53: 874-82. ArticlePubMed
- 36. Broniscer A, Baker SJ, West AN, et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J Clin Oncol 2007; 25: 682-9. ArticlePubMed
- 37. Brodbelt A, Greenberg D, Winters T, et al. Glioblastoma in England: 2007-2011. Eur J Cancer 2015; 51: 533-42. ArticlePubMed
- 38. Lee S, Kim NR, Chung DH, Yee GT, Cho HY. Squash cytology of a dural-based high-grade chondrosarcoma may mimic that of glioblastoma in the central nervous system. Acta Cytol 2015; 59: 219-24. ArticlePubMedPDF
- 39. Lopes MB, Lanzino G, Cloft HJ, Winston DC, Vance ML, Laws ER Jr. Primary fibrosarcoma of the sella unrelated to previous radiation therapy. Mod Pathol 1998; 11: 579-84. PubMed
- 40. Okeda R, Mochizuki T, Terao E, Matsutani M. The origin of intracranial fibrosarcoma. Acta Neuropathol 1980; 52: 223-30. ArticlePubMedPDF
- 41. Franchi A, Santucci M. The contribution of electron microscopy to the characterization of soft tissue fibrosarcomas. Ultrastruct Pathol 2013; 37: 9-14. ArticlePubMed
- 42. Könnecke HK, Rushing EJ, Neidert MC, et al. What’s in a name? Intracranial peripheral primitive neuroectodermal tumors and CNS primitive neuroectodermal tumors are not the same. Strahlenther Onkol 2015 Jul 27 [Epub]. http://dx.doi.org/10.1055/s-0035-1558412.
- 43. Müller K, Diez B, Muggeri A, et al. What’s in a name? Intracranial peripheral primitive neuroectodermal tumors and CNS primitive neuroectodermal tumors are not the same. Strahlenther Onkol 2013; 189: 372-9. ArticlePubMedPDF
- 44. Majumdar K, Batra VV, Tyagi I, Sharma A. Embryonal tumor with abundant neuropil and true rosettes with melanotic (retinal) differentiation. Fetal Pediatr Pathol 2013; 32: 429-36. ArticlePubMed
- 45. Fèvre Montange M, Vasiljevic A, Bergemer Fouquet AM, et al. Histopathologic and ultrastructural features and claudin expression in papillary tumors of the pineal region: a multicenter analysis. Am J Surg Pathol 2012; 36: 916-28. PubMed
- 46. Abela L, Rushing EJ, Ares C, et al. Pediatric papillary tumors of the pineal region: to observe or to treat following gross total resection? Childs Nerv Syst 2013; 29: 307-10. ArticlePubMedPDF
- 47. Perry A, Fuller CE, Judkins AR, Dehner LP, Biegel JA. INI1 expression is retained in composite rhabdoid tumors, including rhabdoid meningiomas. Mod Pathol 2005; 18: 951-8. ArticlePubMedPDF
- 48. Sakata H, Kanamori M, Watanabe M, Kumabe T, Tominaga T. Medulloblastoma demonstrating multipotent differentiation: case report. Brain Tumor Pathol 2008; 25: 39-43. ArticlePubMedPDF
- 49. Weeks DA, Malott RL, Goin L, Mierau GW. Ultrastructural spectrum of medulloblastoma with immunocytochemical correlations. Ultrastruct Pathol 2003; 27: 101-7. ArticlePubMed
- 50. Hassoun J, Gambarelli D, Grisoli F, et al. Central neurocytoma. An electron-microscopic study of two cases. Acta Neuropathol 1982; 56: 151-6. ArticlePubMedPDF
- 51. Iida M, Tsujimoto S, Nakayama H, Yagishita S. Ultrastructural study of neuronal and related tumors in the ventricles. Brain Tumor Pathol 2008; 25: 19-23. ArticlePubMedPDF
- 52. Bouvier-Labit C, Daniel L, Dufour H, Grisoli F, Figarella-Branger D. Papillary glioneuronal tumour: clinicopathological and biochemical study of one case with 7-year follow up. Acta Neuropathol 2000; 99: 321-6. ArticlePubMedPDF
- 53. Miller S, Ward JH, Rogers HA, Lowe J, Grundy RG. Loss of INI1 protein expression defines a subgroup of aggressive central nervous system primitive neuroectodermal tumors. Brain Pathol 2013; 23: 19-27. PubMed
- 54. Judkins AR, Burger PC, Hamilton RL, et al. INI1 protein expression distinguishes atypical teratoid/rhabdoid tumor from choroid plexus carcinoma. J Neuropathol Exp Neurol 2005; 64: 391-7. ArticlePubMed
- 55. Antonelli M, Cenacchi G, Modena P, Morra I, Forni M, Giangaspero F. Ultrastructural evidence of ependymal differentiation in a genetically proven atypical teratoid/rhabdoid tumor. Childs Nerv Syst 2009; 25: 1627-31. ArticlePubMedPDF
- 56. Issidorides MR, Havaki S, Chrysanthou-Piterou M, Arvanitis DL. Ultrastructural identification of protein bodies, cellular markers of human catecholamine neurons, in a temporal lobe ganglioglioma. Ultrastruct Pathol 2000; 24: 399-405. ArticlePubMed
- 57. Kawano N, Yada K, Aihara M, Yagishita S. Oligodendrogliomalike cells (clear cells) in ependymoma. Acta Neuropathol 1983; 62: 141-4. ArticlePubMedPDF
- 58. Biernat W, Liberski PP, Kordek R, Zakrzewski K, Polis L, Budka H. Dysembryoplastic neuroectodermal tumor: an ultrastructural study of six cases. Ultrastruct Pathol 2001; 25: 455-67. ArticlePubMed
- 59. Luse SA. Electron microscopic observations of the central nervous system. J Biophys Biochem Cytol 1956; 2: 531-42. ArticlePubMedPMCPDF
- 60. Zulch KJ, Schmid EE. The ependymoma of the side ventricles at the foramen Monro. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr 1955; 193: 214-28. PubMed
- 61. Mut M, Guler-Tezel G, Lopes MB, Bilginer B, Ziyal I, Ozcan OE. Challenging diagnosis: oligodendroglioma versus extraventricular neurocytoma. Clin Neuropathol 2005; 24: 225-9. PubMed
- 62. Rosner MC, Golomb HM. Ribosome-lamella complex in hairy cell leukemia. Ultrastructure and distribution. Lab Invest 1980; 42: 236-47. PubMed
- 63. Hirose T, Giannini C, Scheithauer BW. Ultrastructural features of pleomorphic xanthoastrocytoma: a comparative study with glioblastoma multiforme. Ultrastruct Pathol 2001; 25: 469-78. ArticlePubMed
- 64. Jain D, Sharma MC, Arora R, Sarkar C, Suri V. Clear cell ependymoma: a mimicker of oligodendroglioma: report of three cases. Neuropathology 2008; 28: 366-71. ArticlePubMed
- 65. Min KW, Scheithauer BW. Clear cell ependymoma: a mimic of oligodendroglioma: clinicopathologic and ultrastructural considerations. Am J Surg Pathol 1997; 21: 820-6. ArticlePubMed
- 66. Sikorska B, Papierz W, Zakrzewki K, Fiks T, Polis L, Liberski PP. Ultrastructural heterogeneity of gangliogliomas. Ultrastruct Pathol 2007; 31: 9-14. ArticlePubMed
- 67. Barnes NP, Pollock JR, Harding B, Hayward RD. Papillary glioneuronal tumour in a 4-year-old. Pediatr Neurosurg 2002; 36: 266-70. ArticlePubMedPDF
- 68. Suh YL, Kim NR, Kim JH, Park SH. Suprasellar chordoid glioma combined with Rathke’s cleft cyst. Pathol Int 2003; 53: 780-5. ArticlePubMed
- 69. Myung JK, Byeon SJ, Kim B, et al. Papillary glioneuronal tumors: a review of clinicopathologic and molecular genetic studies. Am J Surg Pathol 2011; 35: 1794-805. PubMed
- 70. Cykowski MD, Wartchow EP, Mierau GW, Stolzenberg ED, Gumerlock MK, Fung KM. Papillary tumor of the pineal region: ultrastructural study of a case. Ultrastruct Pathol 2012; 36: 68-77. ArticlePubMed
- 71. Castellano-Sanchez AA, Schemankewitz E, Mazewski C, Brat DJ. Pediatric chordoid glioma with chondroid metaplasia. Pediatr Dev Pathol 2001; 4: 564-7. ArticlePubMed
- 72. Nakashima N, Goto K, Tsukidate K, Sobue M, Toida M, Takeuchi J. Choroid plexus papilloma: light and electron microscopic study. Virchows Arch A Pathol Anat Histopathol 1983; 400: 201-11. PubMed
- 73. Tena-Suck ML, Gómez-Amador JL, Ortiz-Plata A, Salina-Lara C, Rembao-Bojórquez D, Vega-Orozco R. Rhabdoid choroid plexus carcinoma: a rare histological type. Arq Neuropsiquiatr 2007; 65: 705-9. ArticlePubMed
- 74. Las Heras F, Pritzker KP. Adult variant of atypical teratoid/rhabdoid tumor: immunohistochemical and ultrastructural confirmation of a rare tumor in the sella tursica. Pathol Res Pract 2010; 206: 788-91. ArticlePubMed
- 75. Jay V, Edwards V, Halliday W, Rutka J, Lau R. “Polyphenotypic” tumors in the central nervous system: problems in nosology and classification. Pediatr Pathol Lab Med 1997; 17: 369-89. ArticlePubMed
- 76. Jeon YK, Jung HW, Park SH. Infratentorial giant cell ependymoma: a rare variant of ependymoma. Pathol Res Pract 2004; 200: 717-25. ArticlePubMed
- 77. Abbott JJ, Amirkhan RH, Hoang MP. Malignant melanoma with a rhabdoid phenotype: histologic, immunohistochemical, and ultrastructural study of a case and review of the literature. Arch Pathol Lab Med 2004; 128: 686-8. ArticlePubMedPDF
- 78. Wyatt-Ashmead J, Kleinschmidt-DeMasters BK, Hill DA, et al. Rhabdoid glioblastoma. Clin Neuropathol 2001; 20: 248-55. PubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
The Continuing Value of Ultrastructural Observation in Central Nervous System Neoplasms in Children

Fig. 1. (A) Meningioma shows closely apposed oval- to spindle-shaped tumor cells having interdigitating cell processes lined by well-formed desmosomes (×8,000). Inset indicates tonofibrils attached to the desmosomal plaques (×30,000). (B) Schwannoma shows abundant reduplicated continuous basal lamina surrounding the interdigitating cell processes (×3,500). Note the extracellular long-spacing collagen (arrow) and high power view of reduplicated basal lamina (inset, ×6,500). (C) Ependymoma reveals spindle cells with numerous cytoplasmic processes filled with bundles of intermediate filaments (×4,000). Inset shows well-formed zonula occludens with surface microvilli (×15,000). (D) Pilomyxoid astrocytoma shows bipolar cells with surface microvilli and cilia in the electron-lucent extracellular space (×9,000). Note some intracellular microlumen. (E) Astrocytoma shows loosely scattered round to oval cells having numerous long cytoplasmic processes filled with bundles of glial filaments and scarce other organelles (×2,500). (F) Glioblastoma shows many pleomorphic spindle and oval cells with numerous cytoplasmic processes containing bundles of intermediate filaments with surface microvilli-like differentiation (×2,500). (G) Medulloblastoma shows that loosely arranged oval-shaped tumor cells project cytoplasmic processes forming rosettes filled with glial filaments and dense core granules indicating neuroglial differentiation (×9,000). (H) Neuroblastoma reveals that closely packed polygonal to spherical tumor cells have numerous, thin, electron-lucent cell processes forming an interlacing meshwork between groups of cell bodies (×9,000). Inset shows longitudinally-oriented 20 nm microtubules (white arrow) and dense core granules (black arrow, ×15,000). (I) Primitive neuroectodermal tumor reveals round to closely apposed oval-shaped tumor cells with abundant cytoplasmic processes containing sparse organelles including glycogen particles, mitochondria, and some microtubules (×8,000).(J) Atypical teratoid and rhabdoid tumor reveals paranuclear aggregates of intermediate filaments (arrow) compressing the heterochromatic nuclei (×15,000). (K) Oligodendroglioma presents tumor cells with uniform round nuclei and sparse cytoplasmic organelles as well as some irregularly shaped cell processes (arrow) containing occasional globoid collections of intermediate filaments (×3,500). (L) Neurocytoma reveals round tumor cells with a moderate amount of cytoplasm and numerous long thin cell processes containing microtubules, few electron dense core granules and secretory vesicles (arrow), and glial intermediate filaments (×8,000). (M) Dysembryoplastic neuroepithelial tumor reveals oligodendroglial-like cells with elongated bulbous cell processes forming a neuropil-like structure filled with intermediate filaments in electron-lucent mucoid extracellular spaces (×7,000).(N) Papillary tumor of the pineal region reveals polygonal-shaped tumor cells having short-villous cell surfaces and cytoplasm having abundant rough endoplasmic reticulums with distended cisternae and vacuoles (×3,500). (O) Chordoid glioma of the third ventricle reveals tumor cells with cytoplasmic processes filled with intermediate glial filaments and surrounded by basal lamina (×7,000). Note the numerous surface microvilli (arrow). (P) Choroid plexus papilloma shows ovoid to polygonal tumor cells arranged in glandular patterns joined by well-formed junctional complexes and a continuous basal lamina (arrow) surrounding these glandular structures (×1,500). Note the apical portion of tumor cells lined by numerous microvilli and cilia composed of 9+2 microtubules.
Fig. 1.
The Continuing Value of Ultrastructural Observation in Central Nervous System Neoplasms in Children

