 E-submission
E-submission
Articles
- Page Path
- HOME > J Pathol Transl Med > Volume 56(3); 2022 > Article
-
Case Study
Adrenal hemangioblastoma -
Joo-Yeon Koo1
 , Kyung-Hwa Lee1, Joon Hyuk Choi2, Ho Seok Chung3, Chan Choi1
, Kyung-Hwa Lee1, Joon Hyuk Choi2, Ho Seok Chung3, Chan Choi1 -
Journal of Pathology and Translational Medicine 2022;56(3):161-166.
DOI: https://doi.org/10.4132/jptm.2021.12.28
Published online: February 28, 2022
1Department of Pathology, Chonnam National University Research Institute of Medical Science, Bio-Medical Sciences Graduate Program (BMSGP), Chonnam National University Hwasun Hospital and Medical School, Hwasun, Korea
2Department of Pathology, Yeungnam University Hospital, Daegu, Korea
3Department of Urology, Chonnam National University Research Institute of Medical Science, Bio-Medical Sciences Graduate Program (BMSGP), Chonnam National University Hwasun Hospital and Medical School, Hwasun, Korea
- Corresponding Author: Chan Choi, MD, PhD, Department of Pathology, Chonnam National University Research Institute of Medical Science, Bio-Medical Sciences Graduate Program (BMSGP), Chonnam National University Hwasun Hospital and Medical School, 322 Seoyang-ro, Hwasun 58128, Korea, Tel: +82-61-379-7071, Fax: +82-61-379-7099, E-mail: cchoi@jnu.ac.kr
© 2022 The Korean Society of Pathologists/The Korean Society for Cytopathology
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (https://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
- Hemangioblastoma (HB) is a rare benign tumor that most commonly occurs in the cerebellum. HB is composed of neoplastic stromal cells and abundant small vessels. However, the exact origin of stromal cells is controversial. Extraneural HBs have been reported in a small series, and peripheral HBs arising in the adrenal gland are extremely rare. Herein, we report a case of sporadic adrenal HB in a 54-year-old woman. The tumor was a well-circumscribed, yellow mass measuring 4.2 cm in diameter. Histologically, the tumor was composed of small blood vessels and vacuolated stromal cells with clear cytoplasm. On immunohistochemical stain, the stromal cells were positive for S-100 protein, neuron-specific enolase, and synaptophysin. The tumor did not reveal mutation of VHL alleles. We herein present a case of HB of the adrenal gland and review of the literature.
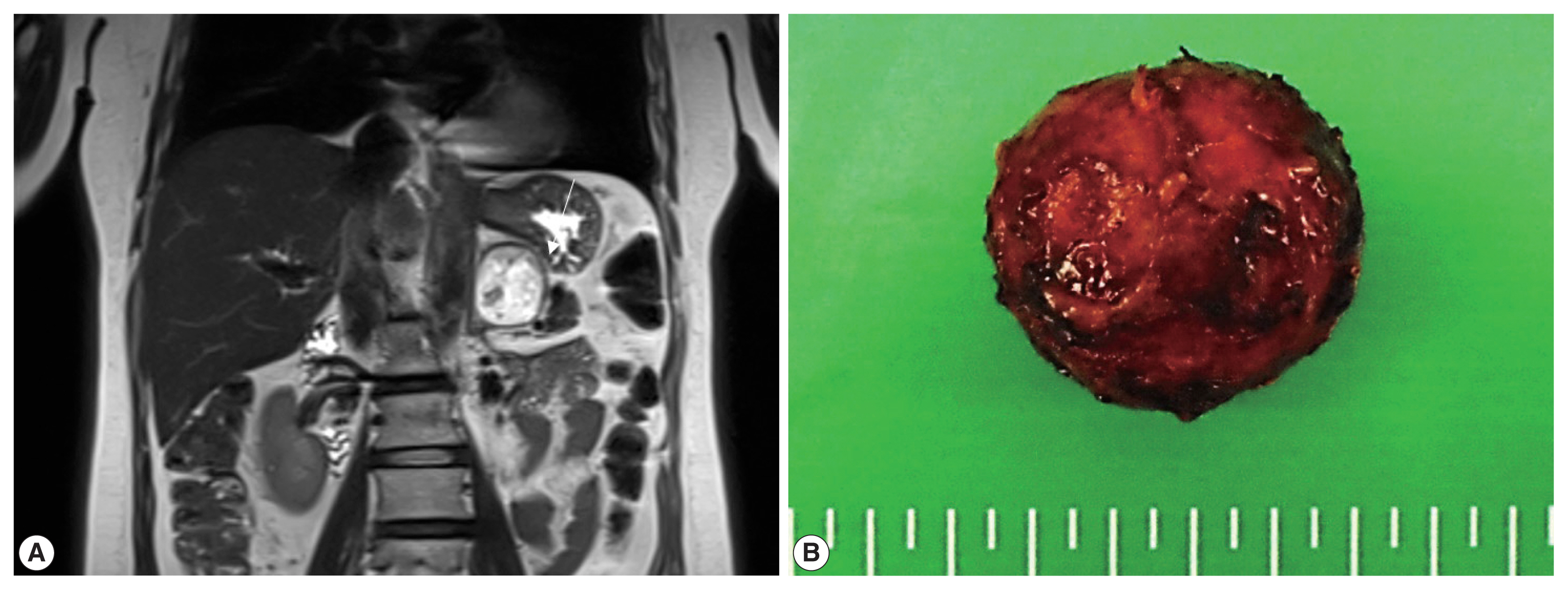
- An adrenal mass was incidentally detected in a 54-year-old woman during a regular medical check-up. She did not have a history of pancreatic tumor, renal tumor, or adrenal tumor. Her family members had no history of such diseases. Laboratory investigations were performed, and 24-hour urinary metanephrine (0.4 mg/day), vanillylmandelic acid (3.3 mg/day), homovanillic acid (4.1 mg/day), and 17-ketosteroid level (16.8 mg/day) were within normal limits. Plasma aldosterone concentration (PAC) was 10.5 ng/dL, plasma renin activity (PRA) was 0.42 ng/mL per hour, and PAC/PRA ratio was 25 ng/dL per ng/mL/hr. Magnetic resonance imaging revealed a 4.2 × 4.2 × 3.6 cm well-defined mass in the left adrenal gland, which was suspected to be a hemangioma (Fig. 1A). The patient underwent partial adrenalectomy. Macroscopic examination revealed that the nodular mass weighed 21 g and displayed a brown-colored cut surface with hemorrhage (Fig. 1B).
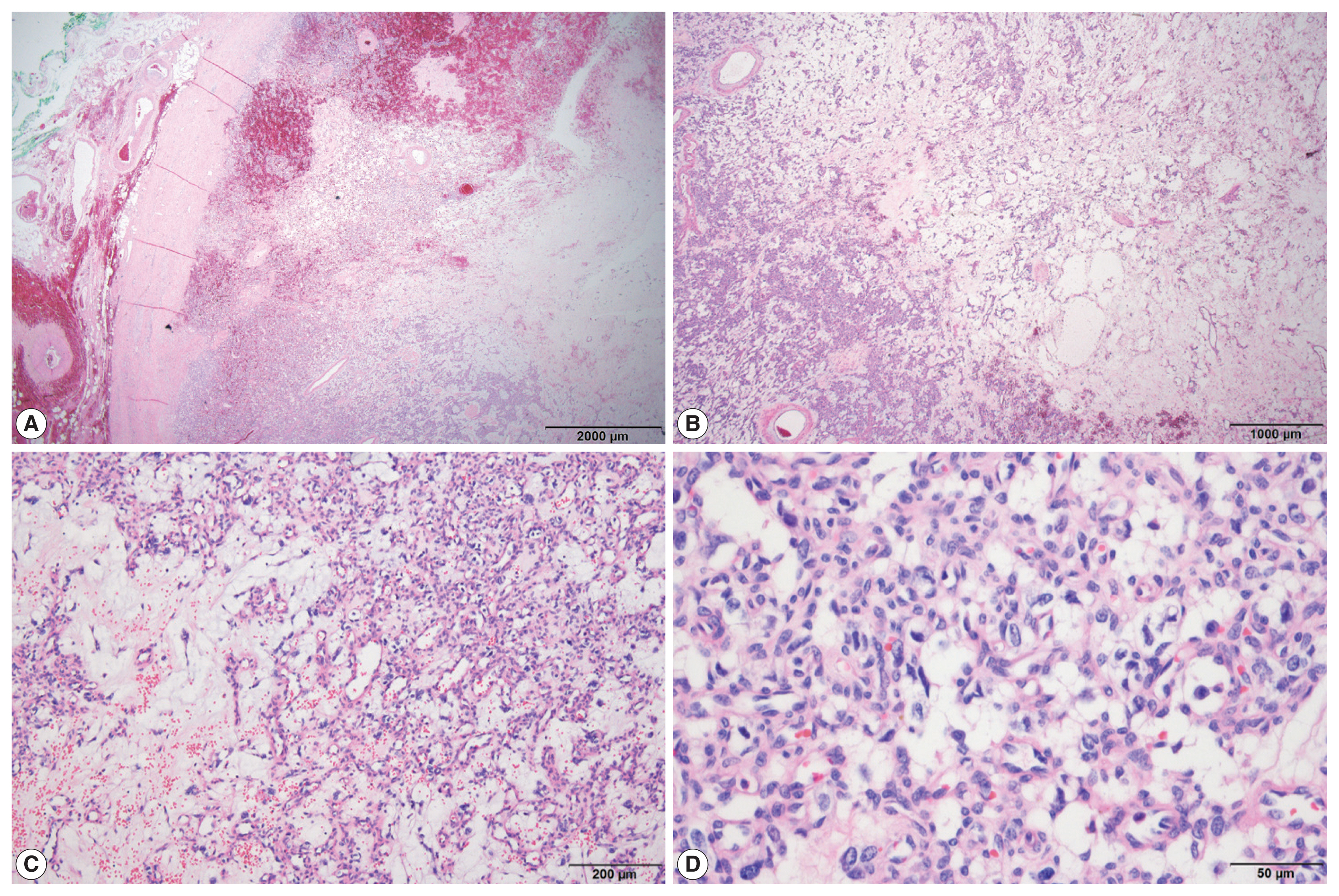
- On histological examination, the tumor was well encapsulated by a fibrous wall. At low magnification, hypocellular and hypercellular area were present. The tumor was composed of abundant small blood vessels admixed with stromal cells. The blood vessels were lined by thin endothelial cells. The stromal cells had clear and vacuolated or pale eosinophilic cytoplasm (Fig. 2). Their nuclei were round to ovoid, with occasional nuclear atypia. There was no evidence of necrosis. On immunohistochemical stain, the stromal cells were positive for S-100 protein, synaptophysin, and neuron-specific enolase (NSE). The endothelial markers, CD34 and CD31, highlighted blood vessels (Fig. 3). The stromal cells were negative for inhibin-α, epithelial membrane antigen (EMA), carbonic anhydrase 9 (CA-9), CD10, glial fibrillary acidic protein (GFAP), GATA3, and chromogranin A. The tumor was diagnosed as HB based on the histological and immunohistochemical findings. The adrenal tissue surrounding the tumor was not submitted.
- Targeted sequencing was performed using the ONCOaccu- Panel (NGeneBio, Seoul, Korea) which covers 323 genes. Capicua transcriptional repressor (CIC), folliculin (FLCN), fms related receptor tyrosine kinase 1 (FLT1), poly (ADP-ribose) polymerase 1 (PARP1), and polybromo 1 (PBRM1) were mutated. However, VHL was not mutated (Table 2). The patient survived with no evidence of tumor recurrence at 1-year follow-up.
CASE REPORT
- HB is a rare benign tumor that generally occurs in the cerebellum in adults. In rare cases, HB has been reported to occur at extracerebellar sites. The unusual anatomical locations and histopathological features of HB may pose a diagnostic challenge. HB has been considered as a vascular neoplasm because of the histological findings of its vascular structure. However, various cell lineages such as glial (GFAP), neural (S-100 protein and NSE), vascular, endothelial (factor XIIIa), mesenchymal (vimentin), epithelial (cytokeratin), and smooth muscle/myofibroblastic (desmin and calponin) have been proposed on the bases of immunohistochemical findings [9–12]. Until now, their cell lineage remains unknown.
- Somatic inactivation of both VHL alleles and the subsequent loss of VHL protein are found in patients with HBs and nonhereditary clear cell renal cell carcinomas (RCCs) [13]. The present case revealed wild-type VHL alleles in targeted sequencing. However, mutations of CIC, FLCN, FLT1, PARP1, and PBRM1 were found (Table 2). CIC is a member of the high-mobility-group-box superfamily of transcriptional repressors. Mutation of CIC is associated with oligodendrogliomas [14], and fusions of CIC with DUX4 (double homeobox 4) and FOXO4 (forkhead box O) genes have been reported in patients with round cell sarcomas [15]. FLCN mutation is associated with Birt-Hogg-Dubé syndrome, which is characterized by benign skin tumors, lung cysts, and kidney lesions [16]. FLT1 encodes a member of the vascular endothelial growth factor receptor family, which plays an important role in angiogenesis and vasculogenesis. PARP1 encodes poly(ADP-ribosyl)transferase, which is involved in cell differentiation, proliferation, recovery from DNA damage, and tumor transformation. PARP1 mutation is associated with Fanconi anemia [17]. PBRM1 encodes a subunit of ATP-dependent chromatin-remodeling complexes, and PBRM1 mutation is associated with clear cell RCC [18] and epithelioid sarcoma [19]. It is unclear whether these mutations are specific for HB, and further studies on these mutations in patients with HBs are needed to clarify this issue.
- HB should be distinguished from their morphological mimickers, such as pheochromocytomas, perivascular epithelioid cell tumors (PEComas), other vascular tumors, and metastatic RCCs. In cases where vacuolated stromal cells are scant and prominent vascular structures are noted, HB may resemble capillary hemangioma. However, capillary hemangioma lacks vacuolated or clear stromal cells, and therefore, careful examination is necessary to make a definitive diagnosis. The distinction of RCC from adrenal HB is considered as one of the most difficult differential diagnostic challenges, because RCC can spread to the adrenal gland, and their histological findings may mimic those of HB. Usually, RCC reveals a nested architecture with thin capillaries. Immunohistochemically, RCC is positive for EMA and paired-box gene 8, and negative for inhibin-alpha and S-100 protein. Furthermore, CA-9 is a marker for clear cell RCC, which is the most common subtype of RCC. Pheochromocytoma may arise in the adrenal medulla. Because of their anatomical location and histological findings of microvascular structure and presence of some vacuolated cells, it should be considered as a differential diagnosis of HB. However, the present case did not display a Zellballen pattern, a characteristic feature of pheochromocytomas. Other histological and immunohistochemical features were not consistent with those of pheochromocytomas. PEComa is a mesenchymal tumor with perivascular epithelioid clear cells that generally develop in the kidney, uterus, and soft tissue. Primary PEComa of the adrenal gland is extremely rare and displays a nested growth pattern of epithelioid cells. PEComa typically expresses both smooth muscle and melanocytic markers such as human melanoma black 45 and Melan-A, whereas HB does not express these markers.
- In summary, adrenal HB is a rare tumor, and its unusual anatomical location and histopathological features may pose a diagnostic challenge. HB should be distinguished from their morphological mimickers, such as pheochromocytoma, PEComa, other vascular tumors, and metastatic RCC. Considering the rarity of adrenal HB, we present a case report of adrenal HB to provide recommendations for its accurate diagnosis and increased awareness of this disease.
DISCUSSION
Ethics Statement
This case report was approved by the IRB of Chonnam Natinal University Hwasun Hospital (CNUHH-2021-240). Informed consent was obtained from the participant included in the study.
Availability of Data and Material
The datasets generated or analyzed during the study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
Author Contributions
Conceptualization: CC. Data curation: JYK, KHL, JHC. Investigation: JYK. Methodology: YJK, KHL, HSC. Supervision: CC. Visualization: JYK. Writing—original draft: JYK. Writing—review & editing: CC, JHC. Approval of final manuscript: all authors.
Conflicts of Interest
J.H.C., a contributing editor of the Journal of Pathology and Translational Medicine, was not involved in the editorial evaluation or decision to publish this article. All remaining authors have declared no conflicts of interest.
Funding Statement
No funding to declare.

| Case | Age (yr)/Sex | Clinical feature | Size/Weight | Microscopic findings | Immunohistochemical or special stain/EM | Association with VHL syndrome | Follow-up |
|---|---|---|---|---|---|---|---|
| Burns et al. (1987) [6] | 23/F | Erythrocytosis (hematocrit, 58%) | 5.0 cm | Abundant thin-walled blood vessels; stromal cells, large cells with vacuolated clear cytoplasm | Stromal cell with fat vacuole | Yes | NA |
| Itoh et. al. (1988) [7] | 49/M | Incidentally found; elevated noradrenalin (187.4 μg/day) and dopamine (2,308 μg/day) in urine | 2.7 cm | Abundant capillary structures; stromal cells, large ovoid | NSE (+), fat stain (+) | No | NA |
| Nonaka et al. (2007) [3] | 30/F | Cerebellar and spinal HB, bilateral adrenal HB | 0.9 cm; 0.5 cm | Focal cavernous vascular spaces; stromal cells, vacuolated large with amphophilic cytoplasm, round to oval nuclei | S-100 protein (+, focal), chromogranin (+), CD31 (−), CD34 (−) | Yes | NA |
| Browning and Parker (2008) [8] | 30/M | Bilateral adrenal HB, spinal and cerebellar HB, pancreatic and renal cysts, cystic renal cell carcinoma (suspicious) | Not identified (12.5 g; 8.3 g) | Abundant capillaries; stromal spindled cells. Stromal cells; large, intimately connected with vascular walls | Chromogranin (+), vimentin (+), S-100 protein (−) | Yes | NA |
| Current case | 54/F | Incidentally found | 4.2 cm | Abundant capillaries; stromal cells, vacuolated or amphophilic cytoplasm | CD34 (−), CD31 (−), inhibin-alpha (−), EMA (−), CK (−), CA-9 (−), CD10 (−), S-100 protein (+), synaptophysin (+), NSE (+) | No | Alive (1 yr) |
- 1. Pakdaman MN, Austin MJ, Bannykh S, Pressman BD. Sporadic hemangioblastoma arising from the infundibulum. J Radiol Case Rep 2017; 11: 1-6. ArticlePubMedPMCPDF
- 2. McGrath FP, Gibney RG, Morris DC, Owen DA, Erb SR. Case report: multiple hepatic and pulmonary haemangioblastomas: a new manifestation of von Hippel-Lindau disease. Clin Radiol 1992; 45: 37-9. ArticlePubMed
- 3. Nonaka D, Rodriguez J, Rosai J. Extraneural hemangioblastoma: a report of 5 cases. Am J Surg Pathol 2007; 31: 1545-51. PubMed
- 4. Wu Y, Wang T, Zhang PP, Yang X, Wang J, Wang CF. Extraneural hemangioblastoma of the kidney: the challenge for clinicopathological diagnosis. J Clin Pathol 2015; 68: 1020-5. ArticlePubMedPMC
- 5. Fanburg-Smith JC, Gyure KA, Michal M, Katz D, Thompson LD. Retroperitoneal peripheral hemangioblastoma: a case report and review of the literature. Ann Diagn Pathol 2000; 4: 81-7. ArticlePubMed
- 6. Burns C, Levine PH, Reichman H, Stock JL. Adrenal hemangioblastoma in Von Hippel-Lindau disease as a cause of secondary erythrocytosis. Am J Med Sci 1987; 293: 119-21. PubMed
- 7. Itoh S, Senda K, Sugita Y, Koike A, Takahashi Y. A case of an adrenal capillary hemangioblastoma. Rinsho Hoshasen 1988; 33: 1047-50. PubMed
- 8. Browning L, Parker A. Bilateral adrenal haemangioblastoma in a patient with von Hippel-Lindau disease. Pathology 2008; 40: 429-31. ArticlePubMed
- 9. Nemes Z. Fibrohistiocytic differentiation in capillary hemangioblastoma. Hum Pathol 1992; 23: 805-10. ArticlePubMed
- 10. Lach B, Gregor A, Rippstein P, Omulecka A. Angiogenic histogenesis of stromal cells in hemangioblastoma: ultrastructural and immunohistochemical study. Ultrastruct Pathol 1999; 23: 299-310. ArticlePubMed
- 11. Barresi V, Vitarelli E, Branca G, Antonelli M, Giangaspero F, Barresi G. Expression of brachyury in hemangioblastoma: potential use in differential diagnosis. Am J Surg Pathol 2012; 36: 1052-7. PubMed
- 12. Doyle LA, Fletcher CD. Peripheral hemangioblastoma: clinicopathologic characterization in a series of 22 cases. Am J Surg Pathol 2014; 38: 119-27. PubMed
- 13. Lee JY, Dong SM, Park WS, et al. Loss of heterozygosity and somatic mutations of the VHL tumor suppressor gene in sporadic cerebellar hemangioblastomas. Cancer Res 1998; 58: 504-8. PubMed
- 14. Cahill DP, Louis DN, Cairncross JG. Molecular background of oligodendroglioma: 1p/19q, IDH, TERT, CIC and FUBP1. CNS Oncol 2015; 4: 287-94. ArticlePubMedPMC
- 15. Yoshimoto T, Tanaka M, Homme M, et al. CIC-DUX4 induces small round cell sarcomas distinct from Ewing sarcoma. Cancer Res 2017; 77: 2927-37. ArticlePubMedPMCPDF
- 16. Schmidt LS, Linehan WM. FLCN: the causative gene for Birt-Hogg-Dube syndrome. Gene 2018; 640: 28-42. ArticlePubMedPMC
- 17. Du W, Amarachintha S, Wilson AF, Pang Q. Hyper-active non-homologous end joining selects for synthetic lethality resistant and pathological Fanconi anemia hematopoietic stem and progenitor cells. Sci Rep 2016; 6: 22167.ArticlePubMedPMCPDF
- 18. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013; 499: 43-9. ArticlePubMedPMCPDF
- 19. Li L, Fan XS, Xia QY, et al. Concurrent loss of INI1, PBRM1, and BRM expression in epithelioid sarcoma: implications for the cocontributions of multiple SWI/SNF complex members to pathogenesis. Hum Pathol 2014; 45: 2247-54. ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Familial Von Hippel–Lindau Disease: A Case Series of Cerebral Hemangioblastomas with MRI, Histopathological, and Genetic Correlations
Claudiu Matei, Ioana Boeras, Dan Orga Dumitriu, Cosmin Mutu, Adriana Popescu, Mihai Gabriel Cucu, Alexandru Calotă-Dobrescu, Bogdan Fetica, Diter Atasie
Life.2025; 15(11): 1649. CrossRef
 PubReader
PubReader ePub Link
ePub Link-
 Cite this Article
Cite this Article
- Cite this Article
-
- Close
- Download Citation
- Close
- Figure
-
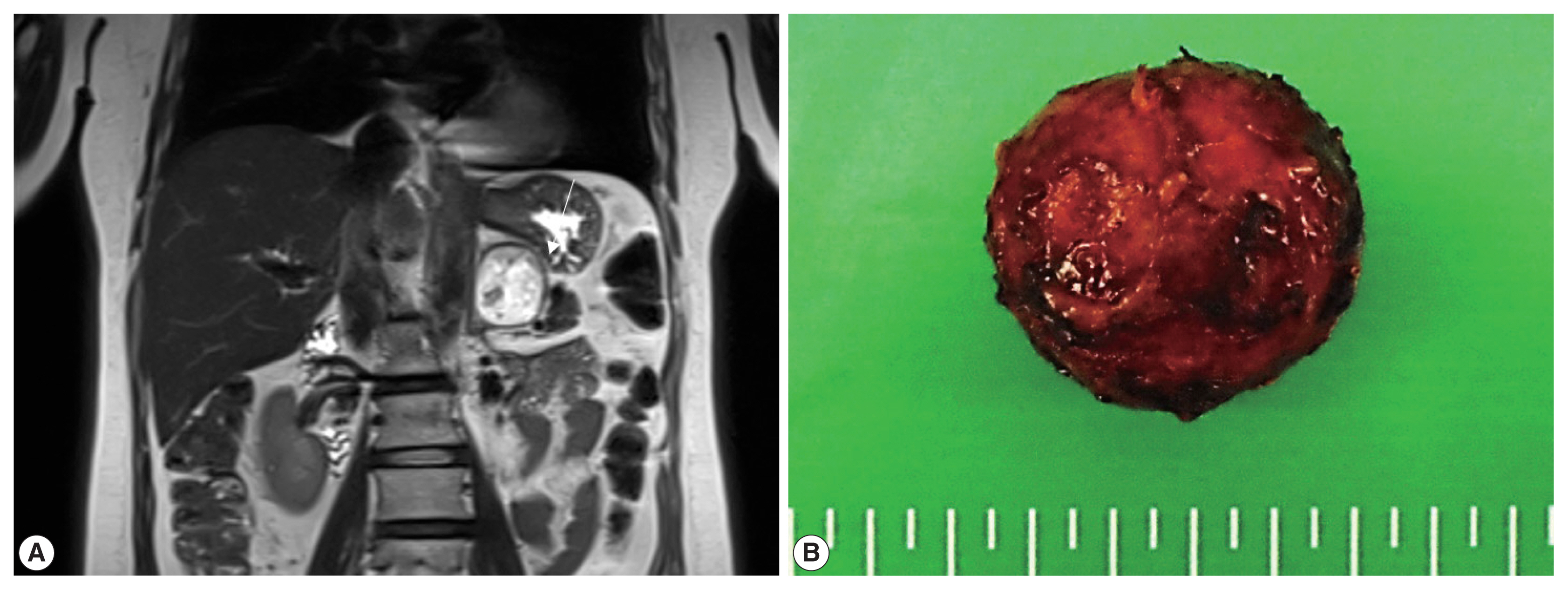
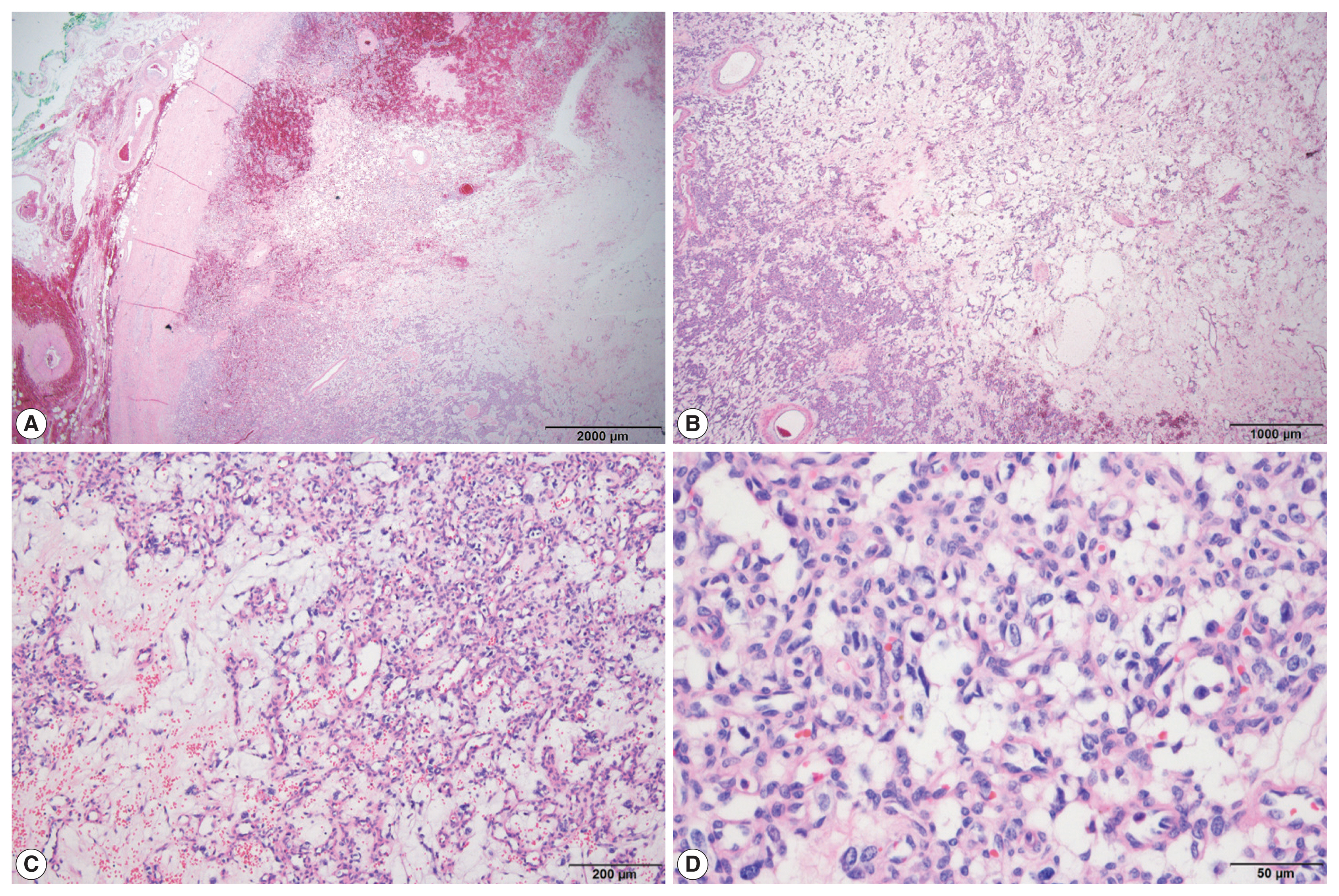
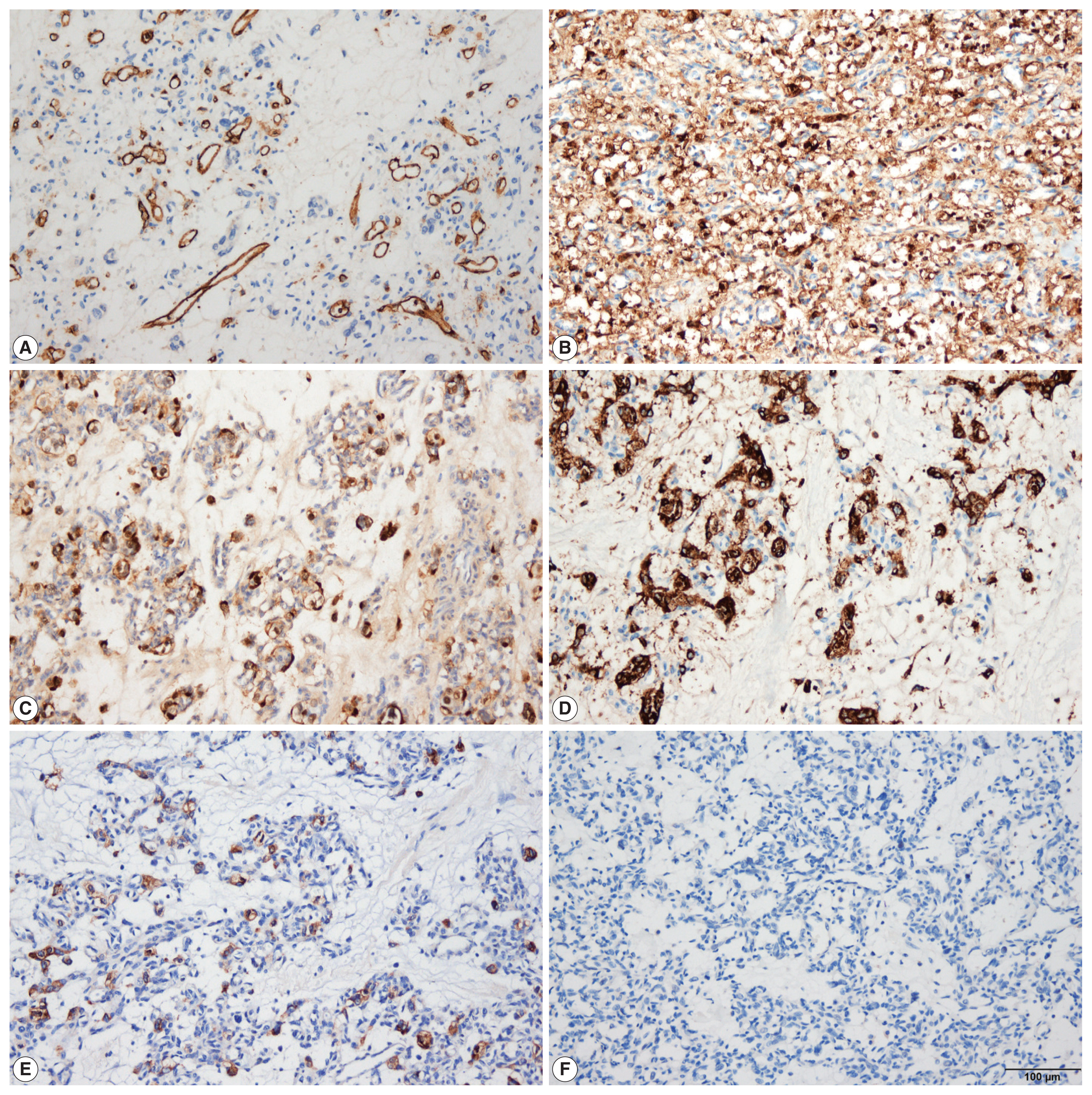



Fig. 1
Fig. 2
Fig. 3
| Case | Age (yr)/Sex | Clinical feature | Size/Weight | Microscopic findings | Immunohistochemical or special stain/EM | Association with VHL syndrome | Follow-up |
|---|---|---|---|---|---|---|---|
| Burns et al. (1987) [ |
23/F | Erythrocytosis (hematocrit, 58%) | 5.0 cm | Abundant thin-walled blood vessels; stromal cells, large cells with vacuolated clear cytoplasm | Stromal cell with fat vacuole | Yes | NA |
| Itoh et. al. (1988) [ |
49/M | Incidentally found; elevated noradrenalin (187.4 μg/day) and dopamine (2,308 μg/day) in urine | 2.7 cm | Abundant capillary structures; stromal cells, large ovoid | NSE (+), fat stain (+) | No | NA |
| Nonaka et al. (2007) [ |
30/F | Cerebellar and spinal HB, bilateral adrenal HB | 0.9 cm; 0.5 cm | Focal cavernous vascular spaces; stromal cells, vacuolated large with amphophilic cytoplasm, round to oval nuclei | S-100 protein (+, focal), chromogranin (+), CD31 (−), CD34 (−) | Yes | NA |
| Browning and Parker (2008) [ |
30/M | Bilateral adrenal HB, spinal and cerebellar HB, pancreatic and renal cysts, cystic renal cell carcinoma (suspicious) | Not identified (12.5 g; 8.3 g) | Abundant capillaries; stromal spindled cells. Stromal cells; large, intimately connected with vascular walls | Chromogranin (+), vimentin (+), S-100 protein (−) | Yes | NA |
| Current case | 54/F | Incidentally found | 4.2 cm | Abundant capillaries; stromal cells, vacuolated or amphophilic cytoplasm | CD34 (−), CD31 (−), inhibin-alpha (−), EMA (−), CK (−), CA-9 (−), CD10 (−), S-100 protein (+), synaptophysin (+), NSE (+) | No | Alive (1 yr) |
| Gene symbol | RefSeq | Amino acid | Nucleotide | No. of variants/Total | VAF (%) |
|---|---|---|---|---|---|
| CIC | NM_015125.3 | Pro911Gln | 2732C>A | 160/365 | 43.84 |
| FLCN | NM_144997.5 | Ser575Leu | 1724C>T | 165/347 | 47.55 |
| FLT1 | NM_002019.4 | Ala673Val | 2018C>T | 348/734 | 47.41 |
| PARP1 | NM_001618.3 | Val991Ala | 2972T>C | 297/555 | 53.51 |
EM, electron microscopy; VHL, von Hippel-Lindau; F, female; NA, not available; M, male; NSE, neuron-specific enolase; HB, hemangioblastoma; EMA, epithelial membrane antigen; CK, cytokeratin; CA-9, carbonic anhydrase 9.
RefSeq, Reference sequence; VAF, variant allele frequency.

